

The Polycomb system sustains promoters in a deep OFF state by limiting pre-initiation complex formation to counteract transcription
- PMID: 39261718
- PMCID: PMC11469961
- DOI: 10.1038/s41556-024-01493-w
The Polycomb system sustains promoters in a deep OFF state by limiting pre-initiation complex formation to counteract transcription
Abstract
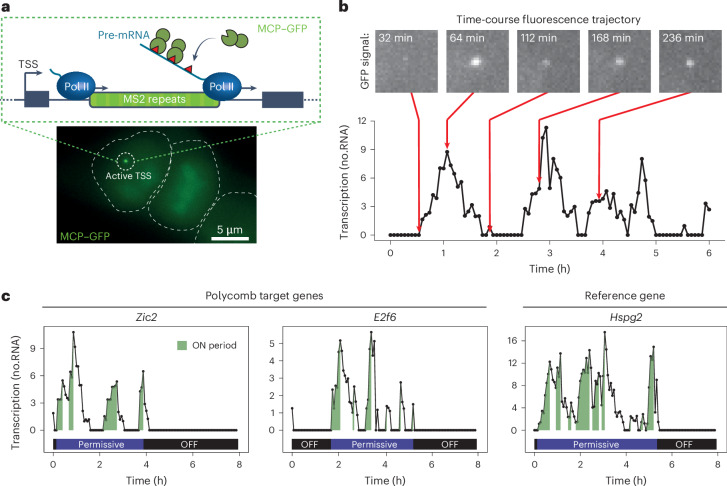
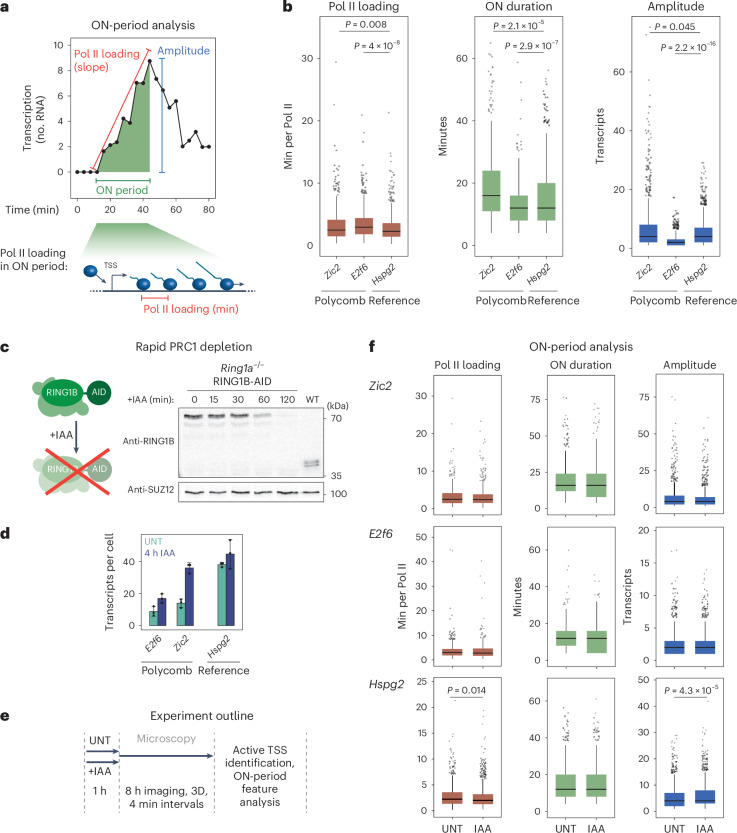
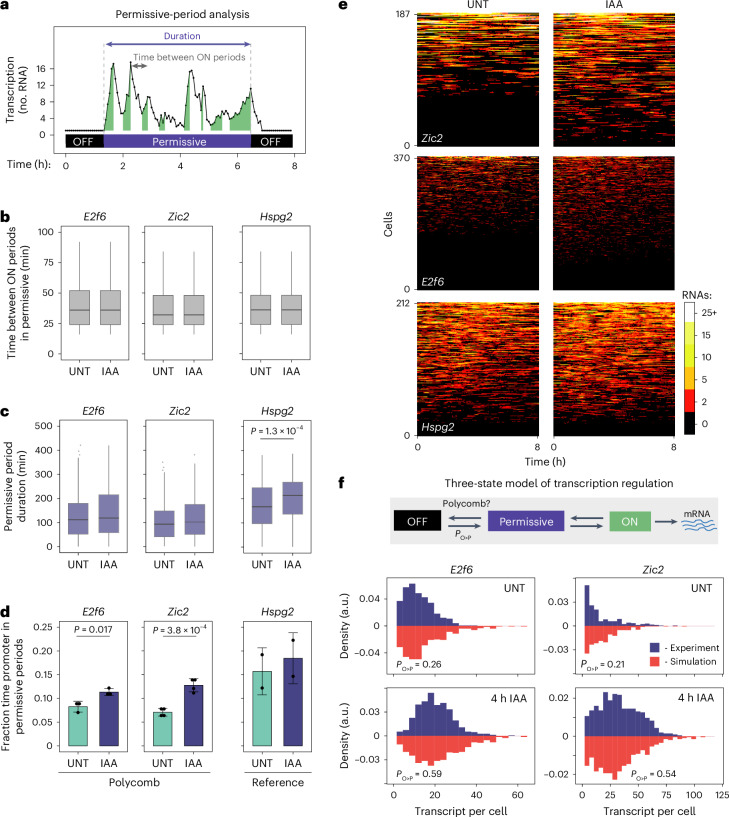
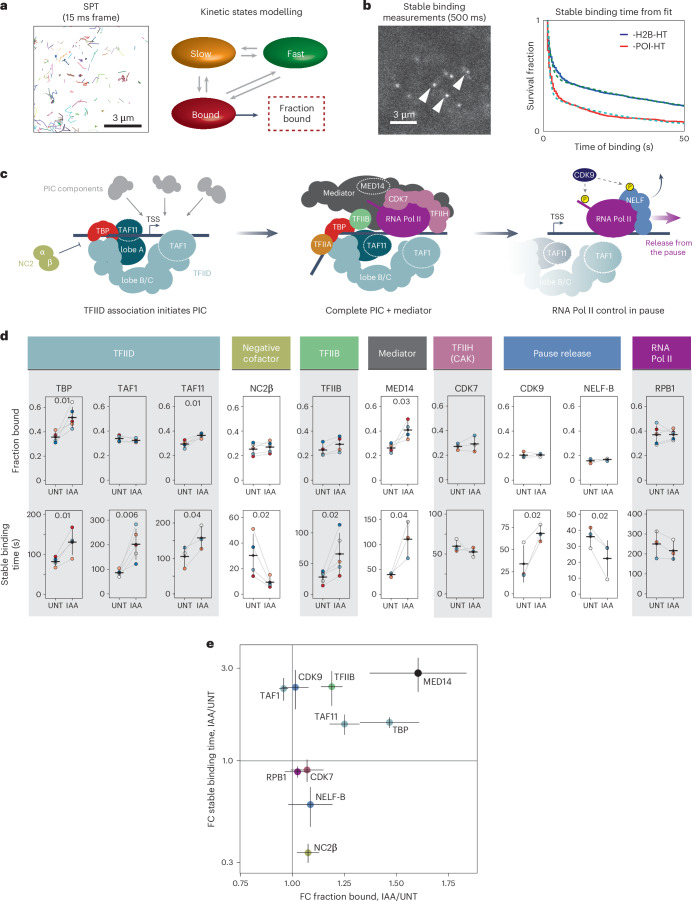
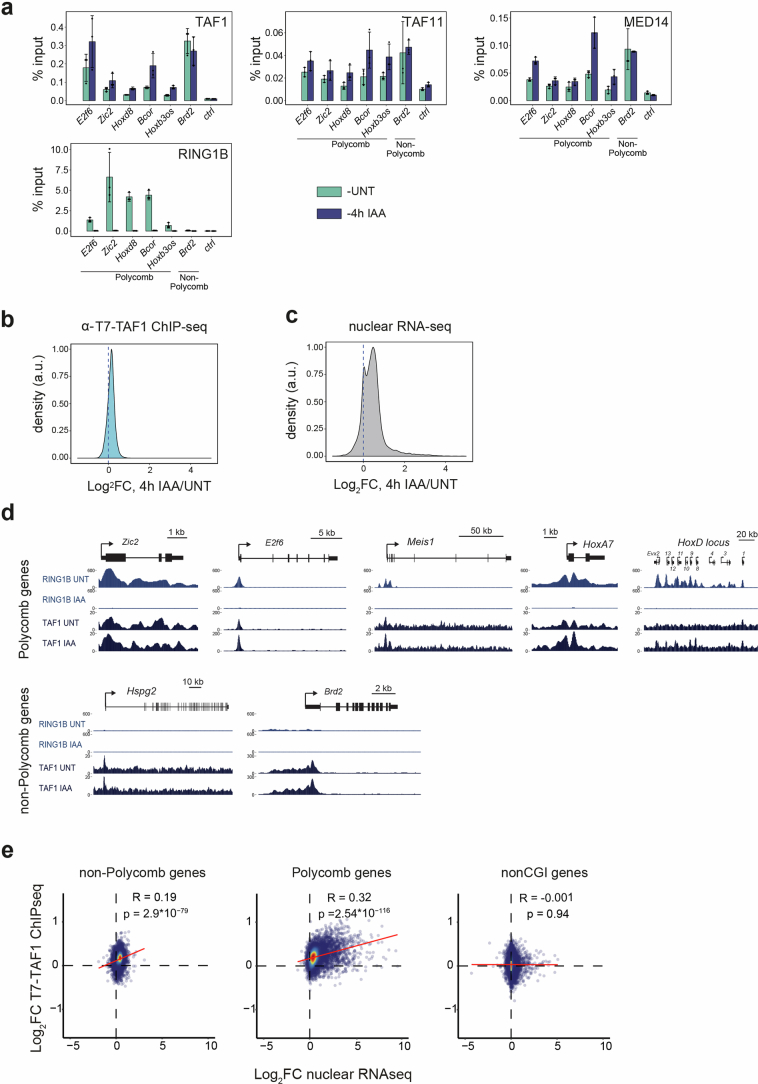
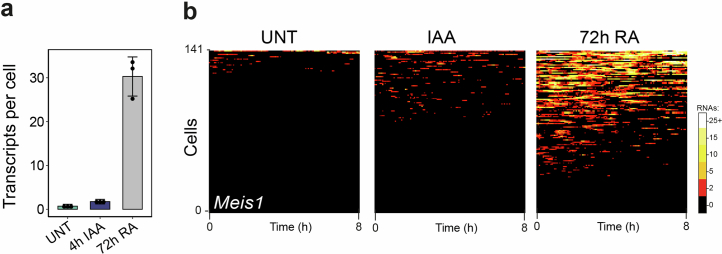
The Polycomb system has fundamental roles in regulating gene expression during mammalian development. However, how it controls transcription to enable gene repression has remained enigmatic. Here, using rapid degron-based depletion coupled with live-cell transcription imaging and single-particle tracking, we show how the Polycomb system controls transcription in single cells. We discover that the Polycomb system is not a constitutive block to transcription but instead sustains a long-lived deep promoter OFF state, which limits the frequency with which the promoter can enter into a transcribing state. We demonstrate that Polycomb sustains this deep promoter OFF state by counteracting the binding of factors that enable early transcription pre-initiation complex formation and show that this is necessary for gene repression. Together, these important discoveries provide a rationale for how the Polycomb system controls transcription and suggests a universal mechanism that could enable the Polycomb system to constrain transcription across diverse cellular contexts.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
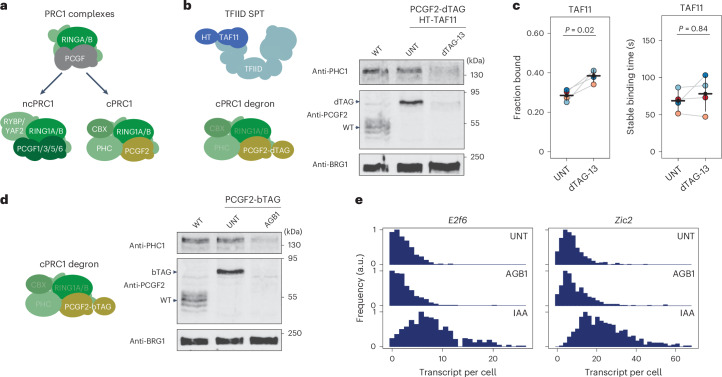
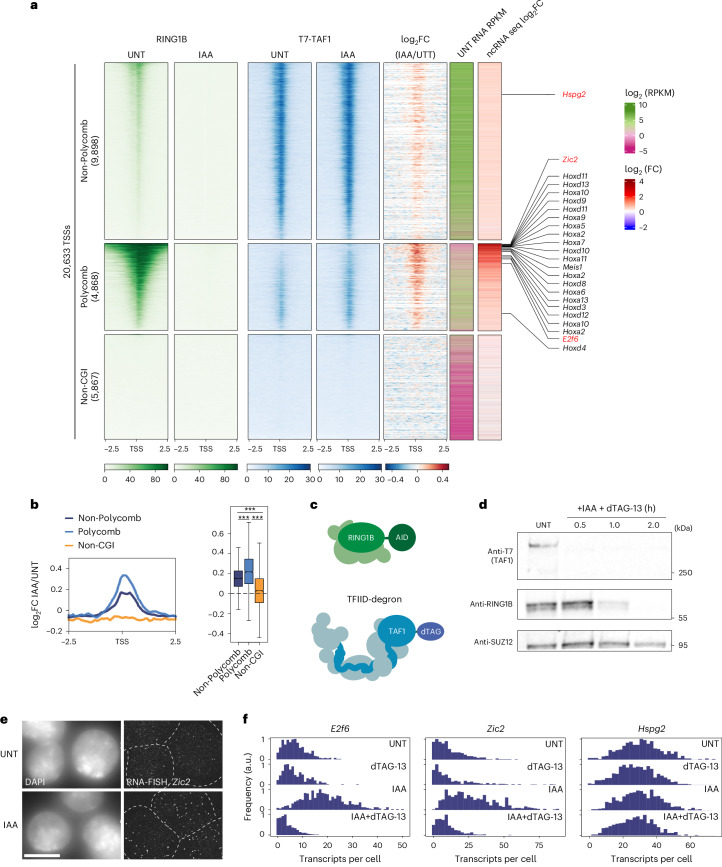
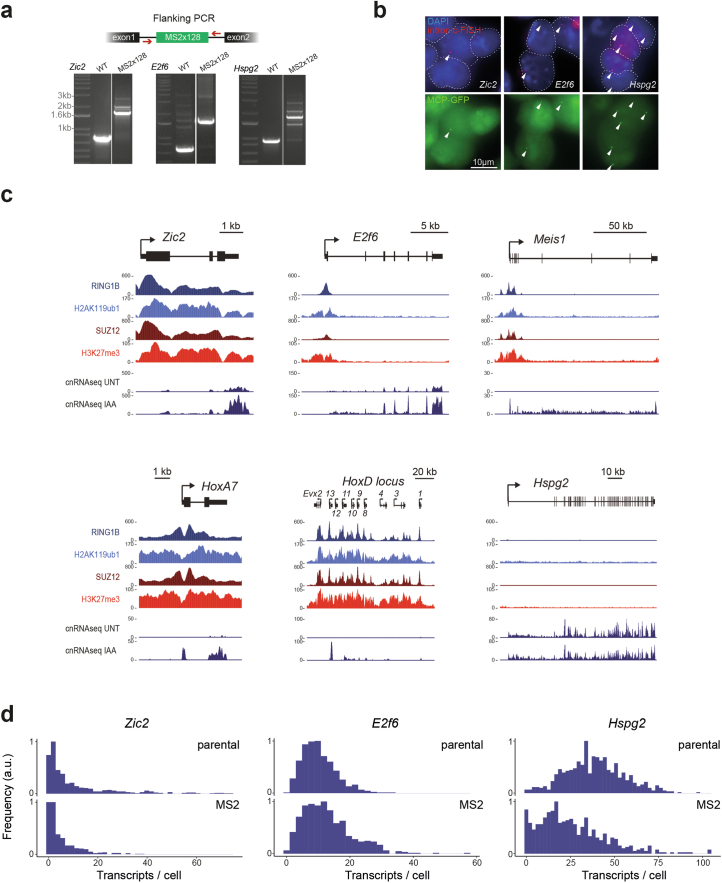
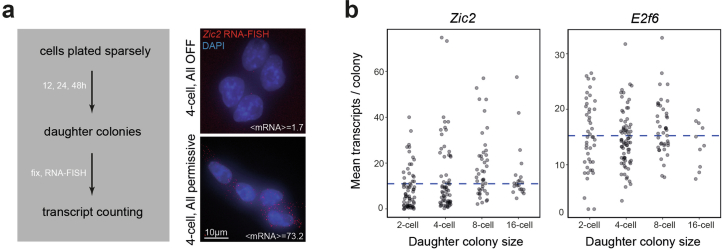
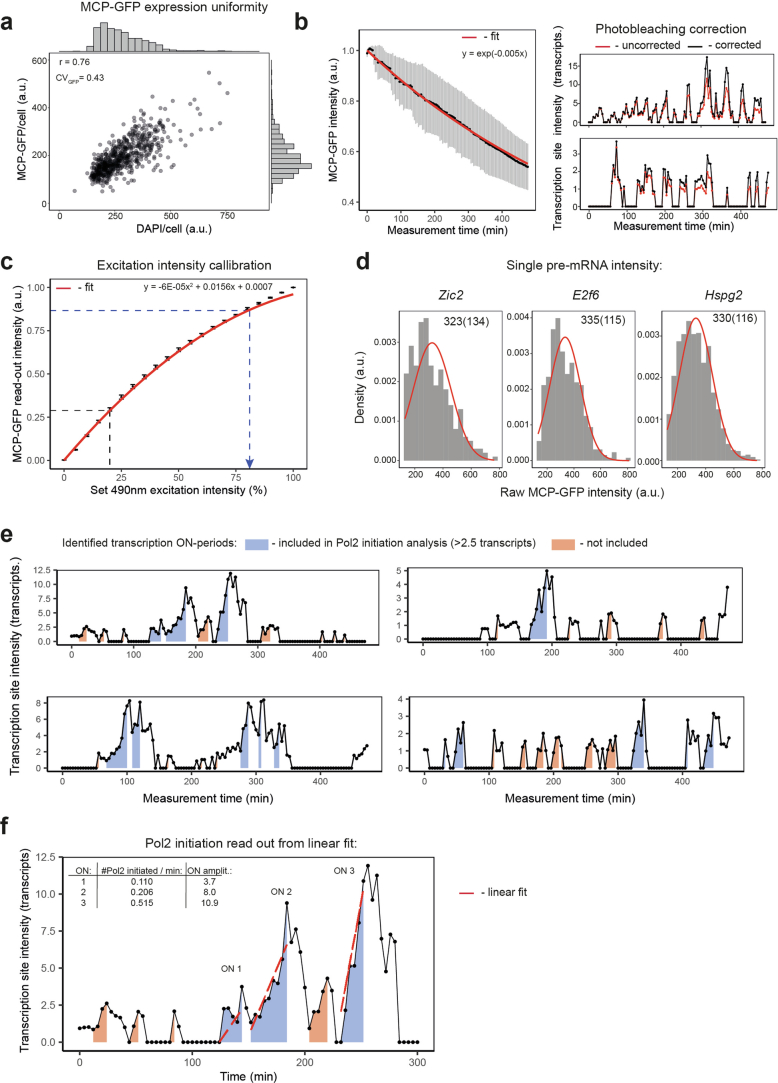
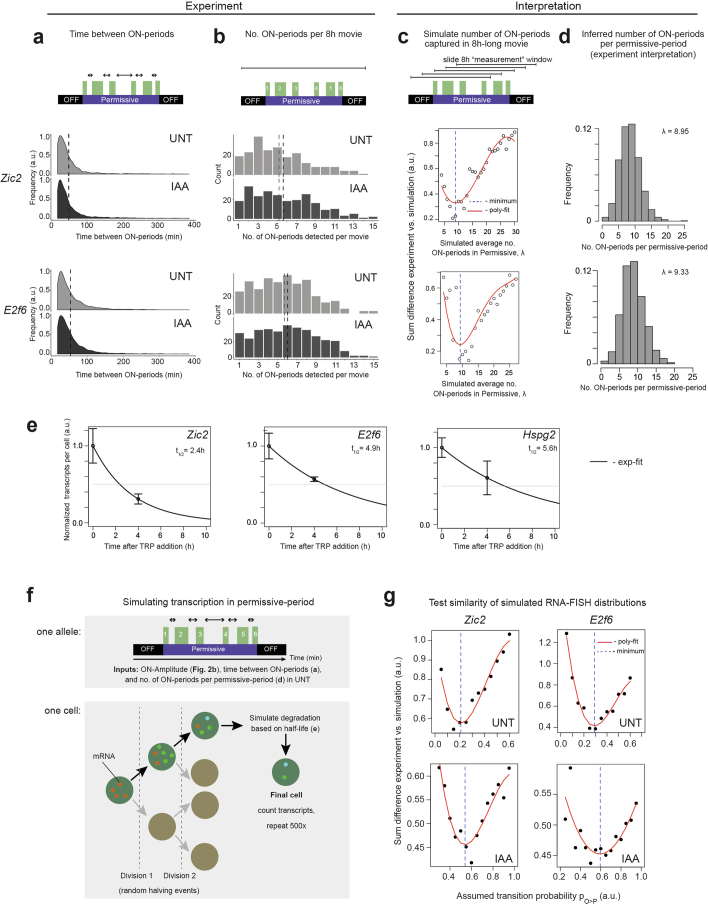
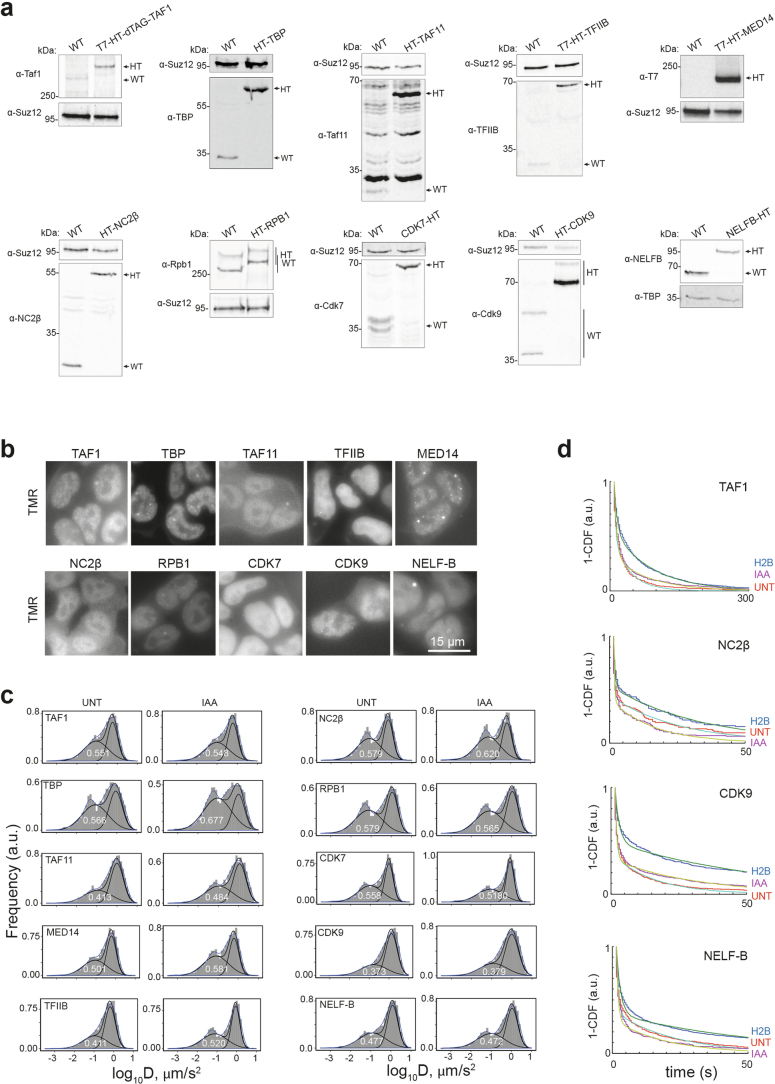
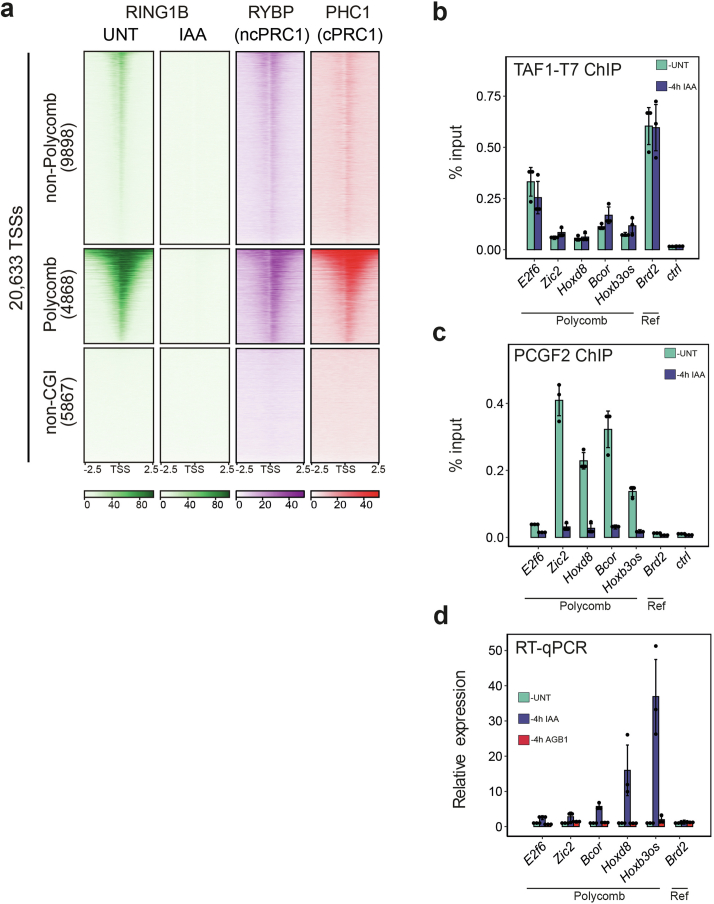
References
-
- Janssen, S. M. & Lorincz, M. C. Interplay between chromatin marks in development and disease. Nat. Rev. Genet.23, 137–153 (2022). - PubMed
-
- Talbert, P. B., Meers, M. P. & Henikoff, S. Old cogs, new tricks: the evolution of gene expression in a chromatin context. Nat. Rev. Genet.20, 283–297 (2019). - PubMed
-
- Kouzarides, T. Chromatin modifications and their function. Cell128, 693–705 (2007). - PubMed
-
- Schuettengruber, B., Bourbon, H. M., Di Croce, L. & Cavalli, G. Genome regulation by polycomb and trithorax: 70 years and counting. Cell171, 34–57 (2017). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
