

Genetic links between ovarian ageing, cancer risk and de novo mutation rates
- PMID: 39261734
- PMCID: PMC11410666
- DOI: 10.1038/s41586-024-07931-x
Genetic links between ovarian ageing, cancer risk and de novo mutation rates
Abstract
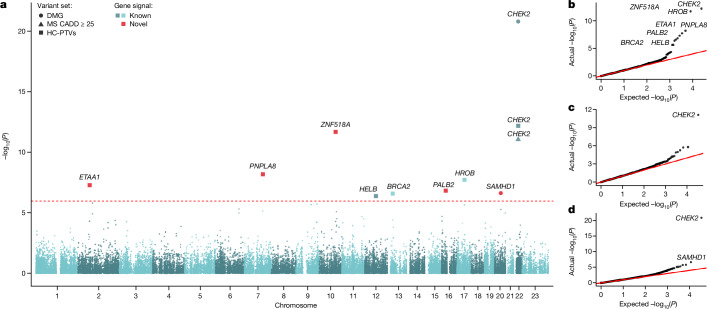
Human genetic studies of common variants have provided substantial insight into the biological mechanisms that govern ovarian ageing1. Here we report analyses of rare protein-coding variants in 106,973 women from the UK Biobank study, implicating genes with effects around five times larger than previously found for common variants (ETAA1, ZNF518A, PNPLA8, PALB2 and SAMHD1). The SAMHD1 association reinforces the link between ovarian ageing and cancer susceptibility1, with damaging germline variants being associated with extended reproductive lifespan and increased all-cause cancer risk in both men and women. Protein-truncating variants in ZNF518A are associated with shorter reproductive lifespan-that is, earlier age at menopause (by 5.61 years) and later age at menarche (by 0.56 years). Finally, using 8,089 sequenced trios from the 100,000 Genomes Project (100kGP), we observe that common genetic variants associated with earlier ovarian ageing associate with an increased rate of maternally derived de novo mutations. Although we were unable to replicate the finding in independent samples from the deCODE study, it is consistent with the expected role of DNA damage response genes in maintaining the genetic integrity of germ cells. This study provides evidence of genetic links between age of menopause and cancer risk.
© 2024. The Author(s).
Conflict of interest statement
J.R.B.P. and E.J.G. are employees of Insmed Innovation UK and hold stock/stock options in Insmed. J.R.B.P. has also received consultancy fees from Hertility Health and WW International and holds research funding from GSK. The other authors declare no competing interests.
Figures










References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
