

Impact and characterization of serial structural variations across humans and great apes
- PMID: 39266513
- PMCID: PMC11393467
- DOI: 10.1038/s41467-024-52027-9
Impact and characterization of serial structural variations across humans and great apes
Abstract
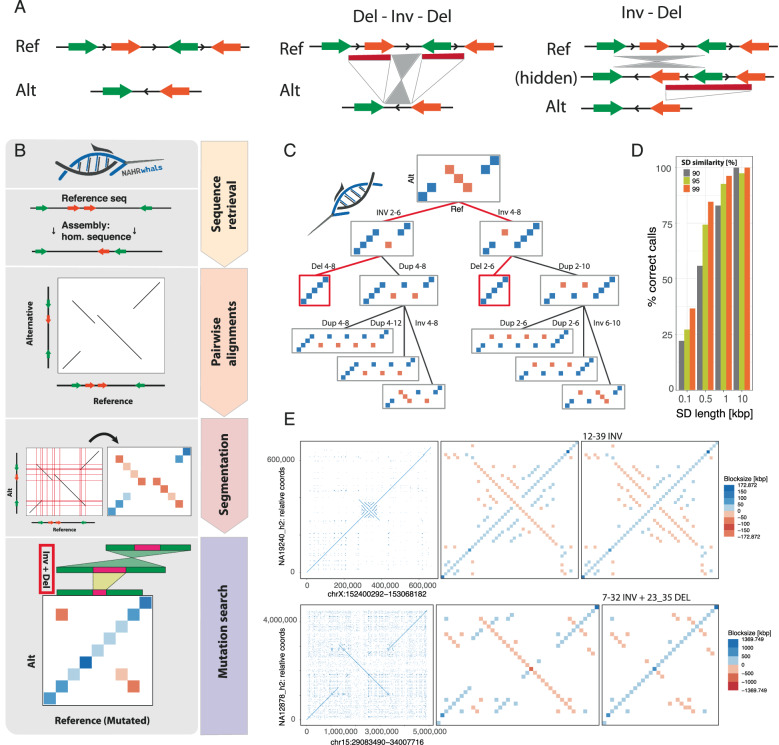
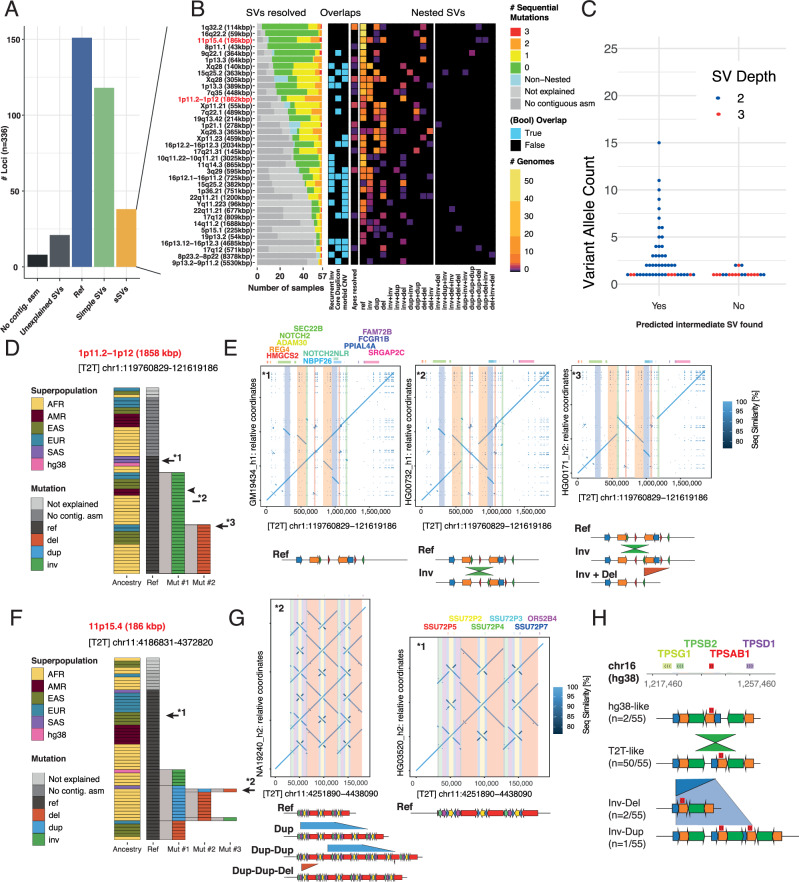
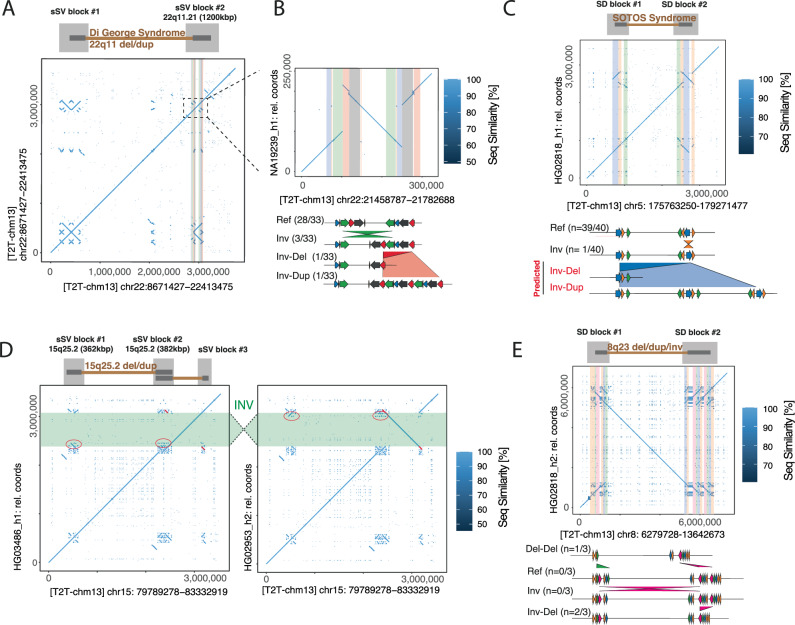
Modern sequencing technology enables the systematic detection of complex structural variation (SV) across genomes. However, extensive DNA rearrangements arising through a series of mutations, a phenomenon we refer to as serial SV (sSV), remain underexplored, posing a challenge for SV discovery. Here, we present NAHRwhals ( https://github.com/WHops/NAHRwhals ), a method to infer repeat-mediated series of SVs in long-read genomic assemblies. Applying NAHRwhals to haplotype-resolved human genomes from 28 individuals reveals 37 sSV loci of various length and complexity. These sSVs explain otherwise cryptic variation in medically relevant regions such as the TPSAB1 gene, 8p23.1, 22q11 and Sotos syndrome regions. Comparisons with great ape assemblies indicate that most human sSVs formed recently, after the human-ape split, and involved non-repeat-mediated processes in addition to non-allelic homologous recombination. NAHRwhals reliably discovers and characterizes sSVs at scale and independent of species, uncovering their genomic abundance and suggesting broader implications for disease.
© 2024. The Author(s).
Conflict of interest statement
J.O.K. has previously disclosed a patent application (no. EP19169090) relevant to Strand-seq. F.J.S receives research support from Illumina, PacBio and ONT. The other authors declare no competing interests.
Figures

References
Publication types
MeSH terms
Grants and funding
- R21 CA259309/CA/NCI NIH HHS/United States
- R01 CA261934/CA/NCI NIH HHS/United States
- U24 HG007497/HG/NHGRI NIH HHS/United States
- U24HG007497/U.S. Department of Health & Human Services | National Institutes of Health (NIH)
- 031L0181A/Bundesministerium für Bildung und Forschung (Federal Ministry of Education and Research)
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
