

SHANK3 depletion leads to ERK signalling overdose and cell death in KRAS-mutant cancers
- PMID: 39266533
- PMCID: PMC11393128
- DOI: 10.1038/s41467-024-52326-1
SHANK3 depletion leads to ERK signalling overdose and cell death in KRAS-mutant cancers
Abstract
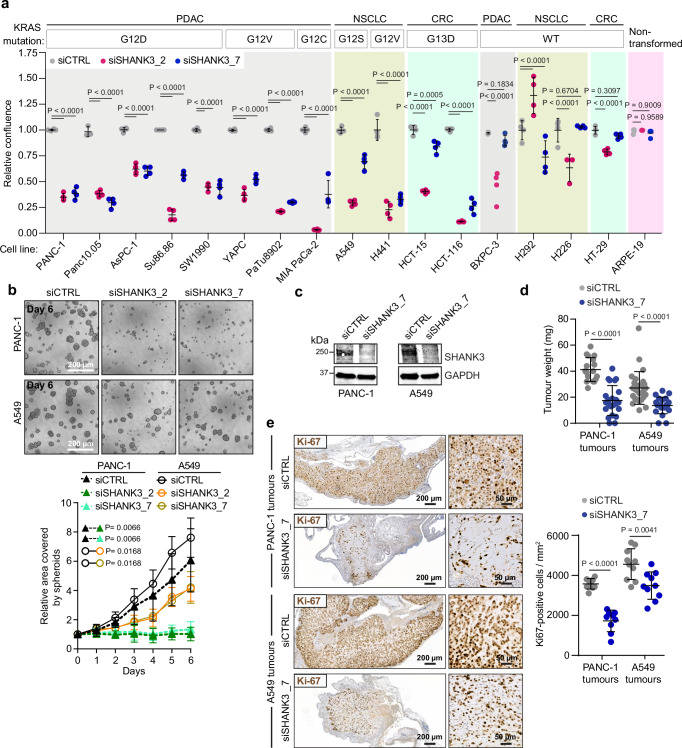
The KRAS oncogene drives many common and highly fatal malignancies. These include pancreatic, lung, and colorectal cancer, where various activating KRAS mutations have made the development of KRAS inhibitors difficult. Here we identify the scaffold protein SH3 and multiple ankyrin repeat domain 3 (SHANK3) as a RAS interactor that binds active KRAS, including mutant forms, competes with RAF and limits oncogenic KRAS downstream signalling, maintaining mitogen-activated protein kinase/extracellular signal-regulated kinase (MAPK/ERK) activity at an optimal level. SHANK3 depletion breaches this threshold, triggering MAPK/ERK signalling hyperactivation and MAPK/ERK-dependent cell death in KRAS-mutant cancers. Targeting this vulnerability through RNA interference or nanobody-mediated disruption of the SHANK3-KRAS interaction constrains tumour growth in vivo in female mice. Thus, inhibition of SHANK3-KRAS interaction represents an alternative strategy for selective killing of KRAS-mutant cancer cells through excessive signalling.
© 2024. The Author(s).
Conflict of interest statement
The authors J.L. and J.I. declare that they have filed patent applications related to these findings (Therapy of Ras-Dependent Cancers, WO/2021/160937; Nanobodies specifically binding to SH3 and multiple ankyrin repeat domains 3 (SHANK3), WO/2023/021181). All other authors declare no competing interests.
Figures





References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
