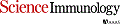CD4+ T cells with convergent TCR recombination reprogram stroma and halt tumor progression in adoptive therapy
- PMID: 39270007
- PMCID: PMC11560124
- DOI: 10.1126/sciimmunol.adp6529
CD4+ T cells with convergent TCR recombination reprogram stroma and halt tumor progression in adoptive therapy
Abstract
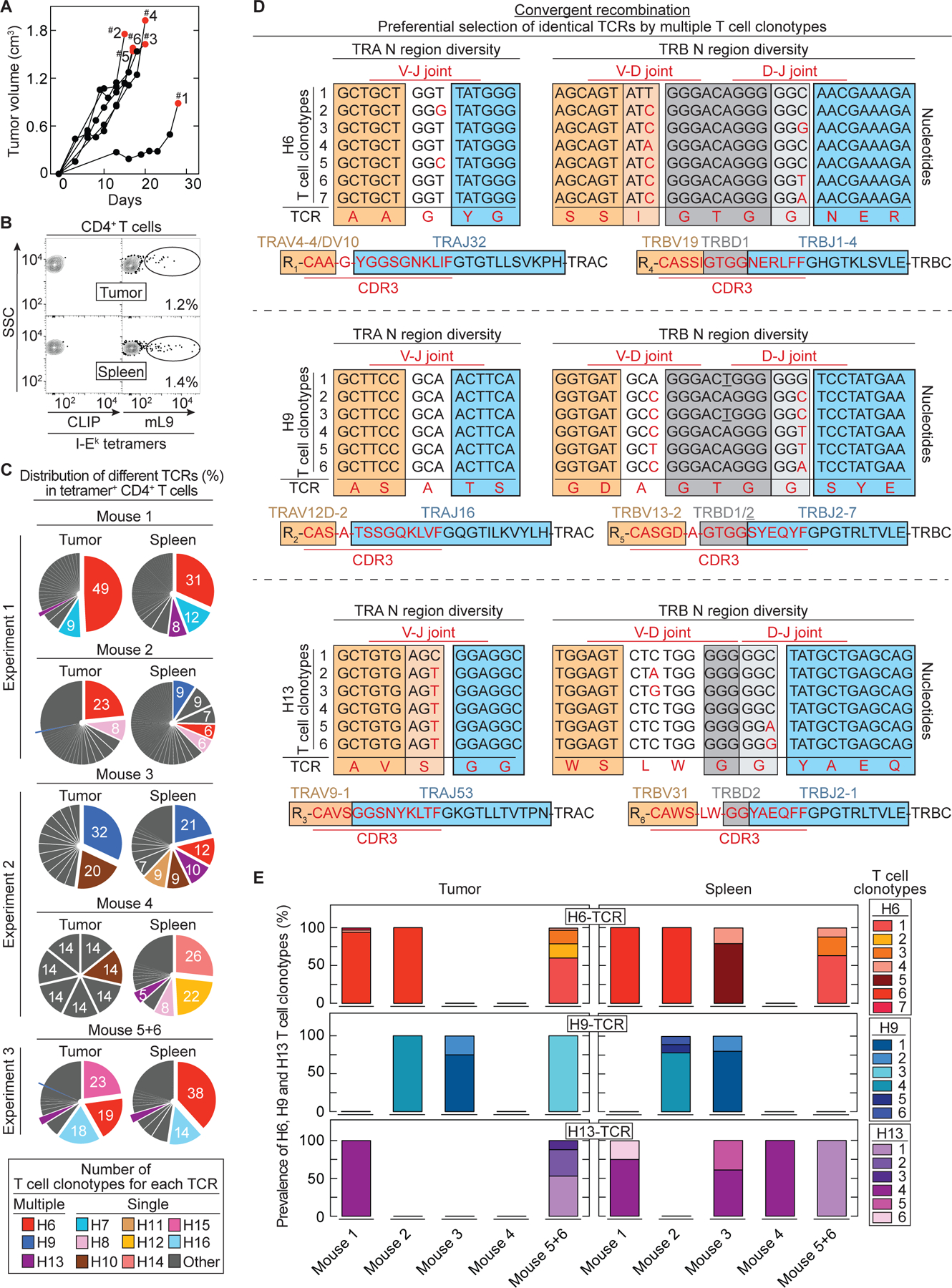
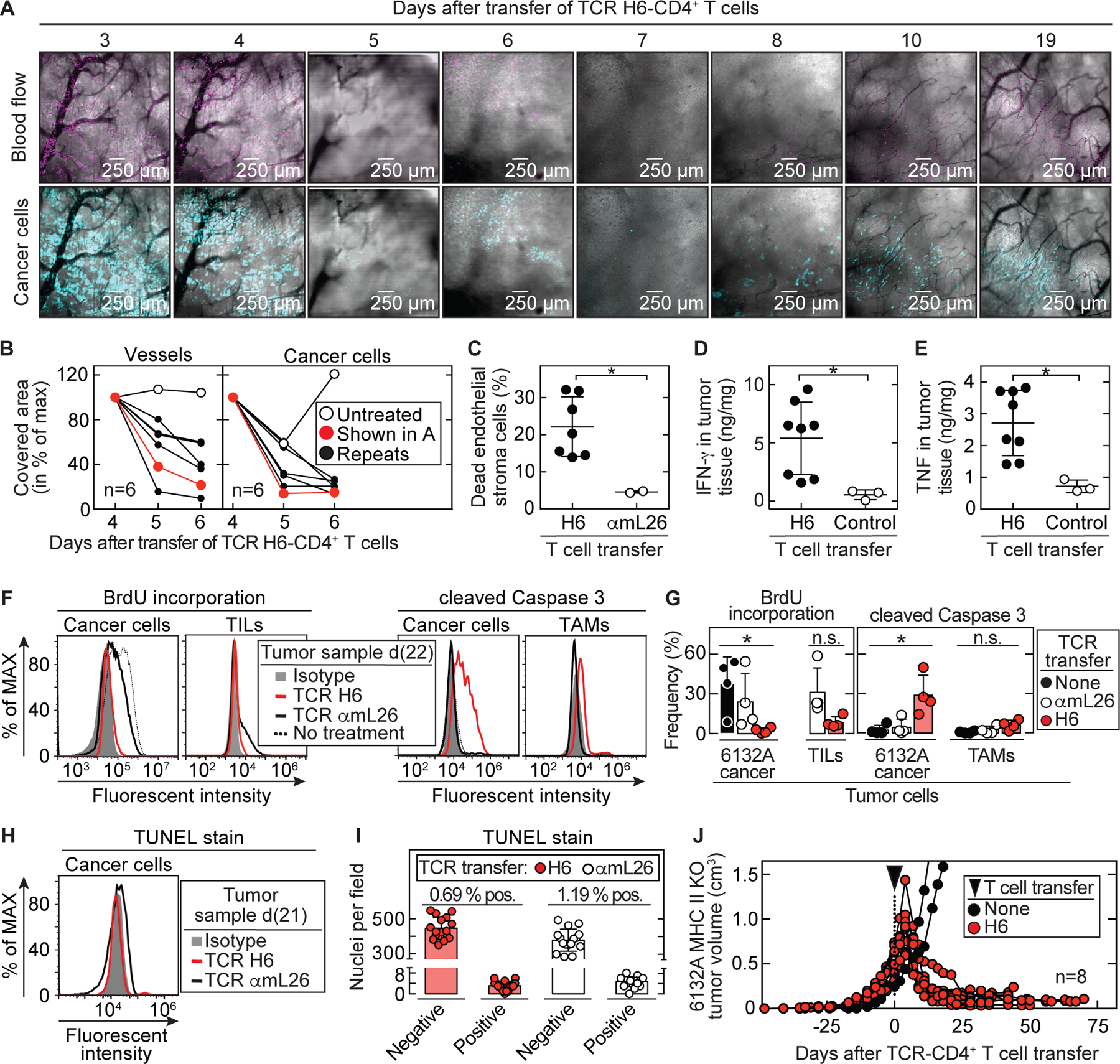
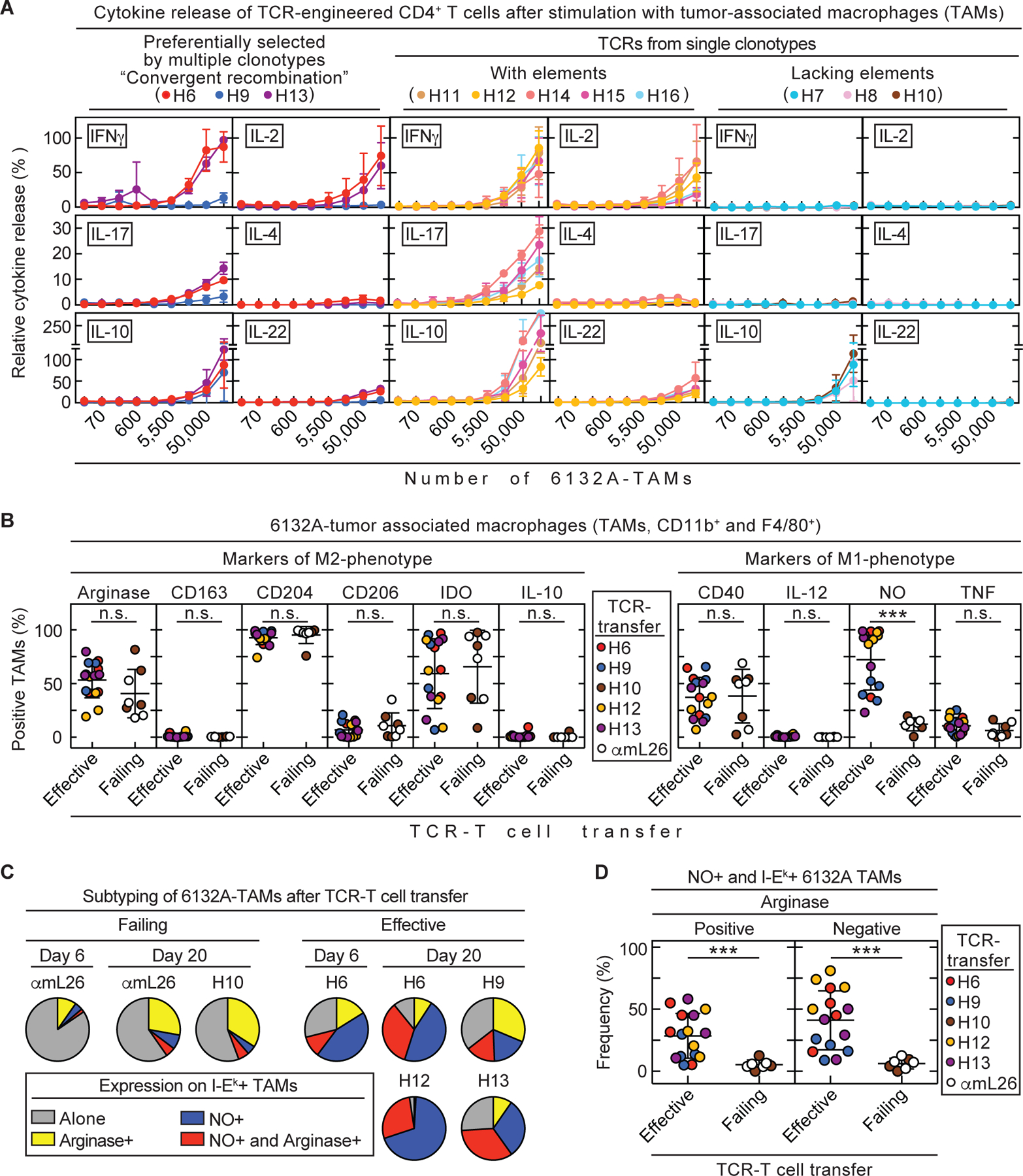
Cancers eventually kill hosts even when infiltrated by cancer-specific T cells. We examined whether cancer-specific T cell receptors of CD4+ T cells (CD4TCRs) from tumor-bearing hosts can be exploited for adoptive TCR therapy. We focused on CD4TCRs targeting an autochthonous mutant neoantigen that is only presented by stroma surrounding the MHC class II-negative cancer cells. The 11 most common tetramer-sorted CD4TCRs were tested using TCR-engineered CD4+ T cells. Three TCRs were characterized by convergent recombination for which multiple T cell clonotypes differed in their nucleotide sequences but encoded identical TCR α and β chains. These preferentially selected TCRs destroyed tumors equally well and halted progression through reprogramming of the tumor stroma. TCRs represented by single T cell clonotypes were similarly effective only if they shared CDR elements with preferentially selected TCRs in both α and β chains. Selecting candidate TCRs on the basis of these characteristics can help identify TCRs that are potentially therapeutically effective.
Conflict of interest statement
Figures

References
-
- Monach PA, Meredith SC, Siegel CT, Schreiber H, A unique tumor antigen produced by a single amino acid substitution. Immunity 2, 45–59 (1995). - PubMed
-
- Wölfel T, Hauer M, Schneider J, Serrano M, Wölfel C, Klehmann-Hieb E, De Plaen E, Hankeln T, Meyer zum Buschenfelde KH, Beach D, A p16INK4a-insensitive CDK4 mutant targeted by cytolytic T lymphocytes in a human melanoma. Science 269, 1281–1284 (1995). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials