

Combining Data Independent Acquisition With Spike-In SILAC (DIA-SiS) Improves Proteome Coverage and Quantification
- PMID: 39271013
- PMCID: PMC11795695
- DOI: 10.1016/j.mcpro.2024.100839
Combining Data Independent Acquisition With Spike-In SILAC (DIA-SiS) Improves Proteome Coverage and Quantification
Abstract
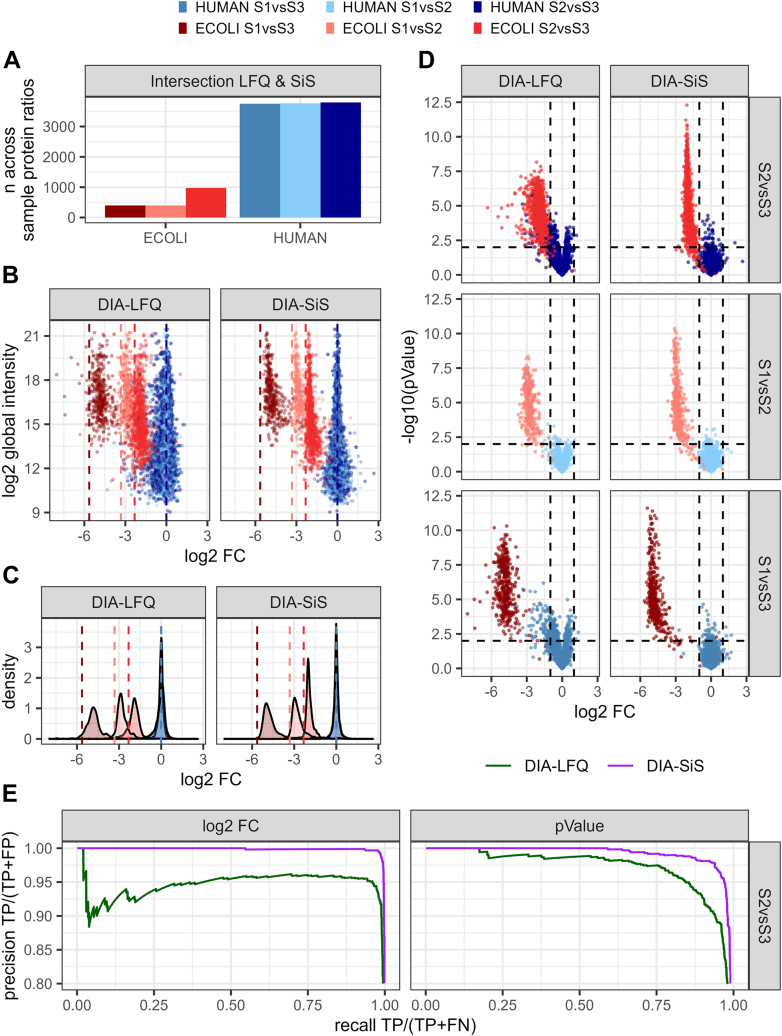
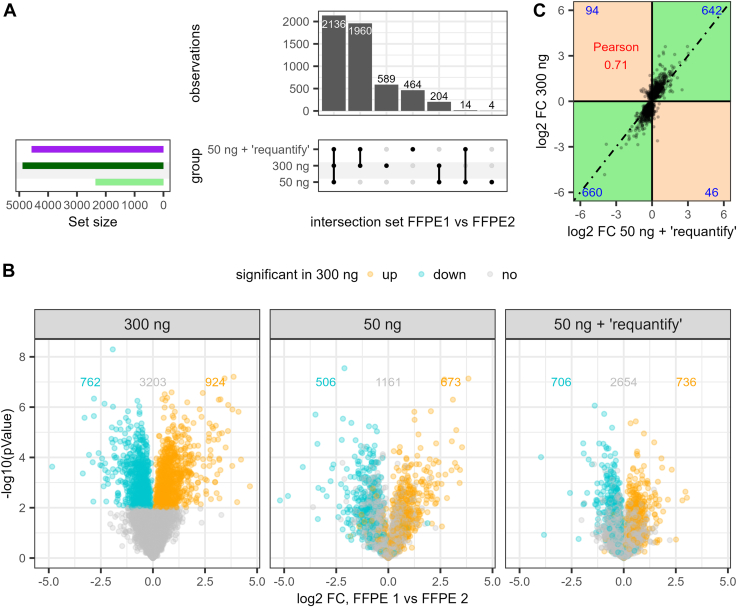
Data-independent acquisition (DIA) is increasingly preferred over data-dependent acquisition due to its higher throughput and fewer missing values. Whereas data-dependent acquisition often uses stable isotope labeling to improve quantification, DIA mostly relies on label-free approaches. Efforts to integrate DIA with isotope labeling include chemical methods like mass differential tags for relative and absolute quantification and dimethyl labeling, which, while effective, complicate sample preparation. Stable isotope labeling by amino acids in cell culture (SILAC) achieves high labeling efficiency through the metabolic incorporation of heavy labels into proteins in vivo. However, the need for metabolic incorporation limits the direct use in clinical scenarios and certain high-throughput experiments. Spike-in SILAC (SiS) methods use an externally generated heavy sample as an internal reference, enabling SILAC-based quantification even for samples that cannot be directly labeled. Here, we combine DIA-SiS, leveraging the robust quantification of SILAC without the complexities associated with chemical labeling. We developed DIA-SiS and rigorously assessed its performance with mixed-species benchmark samples on bulk and single cell-like amount level. We demonstrate that DIA-SiS substantially improves proteome coverage and quantification compared to label-free approaches and reduces incorrectly quantified proteins. Additionally, DIA-SiS proves effective in analyzing proteins in low-input formalin-fixed paraffin-embedded tissue sections. DIA-SiS combines the precision of stable isotope-based quantification with the simplicity of label-free sample preparation, facilitating simple, accurate, and comprehensive proteome profiling.
Keywords: DIA; SILAC; data independent acquisition; multispecies benchmark; spike-in.
Copyright © 2024 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Conflict of interest P. M. and M. S. are Editorial Board Members/Editor-in-Chief/Associate Editors/Guest Editors for Molecular and Cellular Proteomics and were not involved in the editorial review or the decision to publish this article. The other authors declare no competing interests.
Figures




Similar articles
-
MdFDIA: A Mass Defect Based Four-Plex Data-Independent Acquisition Strategy for Proteome Quantification.Anal Chem. 2017 Oct 3;89(19):10248-10255. doi: 10.1021/acs.analchem.7b01635. Epub 2017 Sep 19. Anal Chem. 2017. PMID: 28872844
-
DIA-SIFT: A Precursor and Product Ion Filter for Accurate Stable Isotope Data-Independent Acquisition Proteomics.Anal Chem. 2018 Aug 7;90(15):8722-8726. doi: 10.1021/acs.analchem.8b01618. Epub 2018 Jul 19. Anal Chem. 2018. PMID: 29989796 Free PMC article.
-
Improved SILAC Quantification with Data-Independent Acquisition to Investigate Bortezomib-Induced Protein Degradation.J Proteome Res. 2021 Apr 2;20(4):1918-1927. doi: 10.1021/acs.jproteome.0c00938. Epub 2021 Mar 25. J Proteome Res. 2021. PMID: 33764077 Free PMC article.
-
Stable Isotope Labeling by Amino Acids in Cell Culture (SILAC) for Quantitative Proteomics.Adv Exp Med Biol. 2019;1140:531-539. doi: 10.1007/978-3-030-15950-4_31. Adv Exp Med Biol. 2019. PMID: 31347069 Review.
-
Super-SILAC: current trends and future perspectives.Expert Rev Proteomics. 2015 Feb;12(1):13-9. doi: 10.1586/14789450.2015.982538. Epub 2014 Nov 18. Expert Rev Proteomics. 2015. PMID: 25404501 Review.
Cited by
-
Single-nucleus proteomics identifies regulators of protein transport.bioRxiv [Preprint]. 2024 Jun 18:2024.06.17.599449. doi: 10.1101/2024.06.17.599449. bioRxiv. 2024. PMID: 38948785 Free PMC article. Preprint.
-
Spike-in enhanced phosphoproteomics uncovers synergistic signaling responses to MEK inhibition in colon cancer cells.Nat Commun. 2025 May 27;16(1):4884. doi: 10.1038/s41467-025-59404-y. Nat Commun. 2025. PMID: 40419504 Free PMC article.
-
A robust multiplex-DIA workflow profiles protein turnover regulations associated with cisplatin resistance and aneuploidy.Nat Commun. 2025 May 30;16(1):5034. doi: 10.1038/s41467-025-60319-x. Nat Commun. 2025. PMID: 40447611 Free PMC article.
-
A Comprehensive and Robust Multiplex-DIA Workflow Profiles Protein Turnover Regulations Associated with Cisplatin Resistance.bioRxiv [Preprint]. 2024 Oct 31:2024.10.28.620709. doi: 10.1101/2024.10.28.620709. bioRxiv. 2024. Update in: Nat Commun. 2025 May 30;16(1):5034. doi: 10.1038/s41467-025-60319-x. PMID: 39554001 Free PMC article. Updated. Preprint.
References
-
- Doerr A. DIA mass spectrometry. Nat. Methods. 2015;12:35.
-
- Krasny L., Huang P.H. Data-independent acquisition mass spectrometry (DIA-MS) for proteomic applications in oncology. Mol. Omics. 2021;17:29–42. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
