

Tissue-resident memory T cells in epicardial adipose tissue comprise transcriptionally distinct subsets that are modulated in atrial fibrillation
- PMID: 39271815
- PMCID: PMC11399095
- DOI: 10.1038/s44161-024-00532-x
Tissue-resident memory T cells in epicardial adipose tissue comprise transcriptionally distinct subsets that are modulated in atrial fibrillation
Abstract
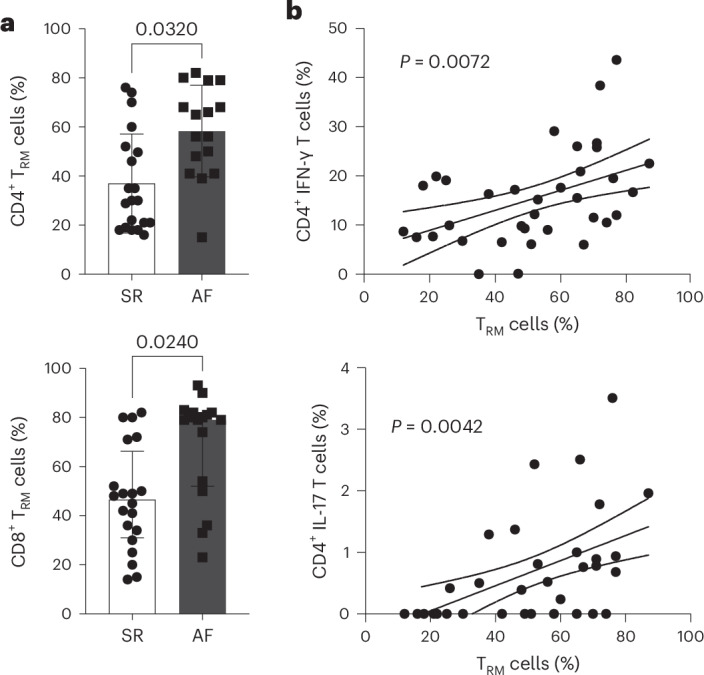
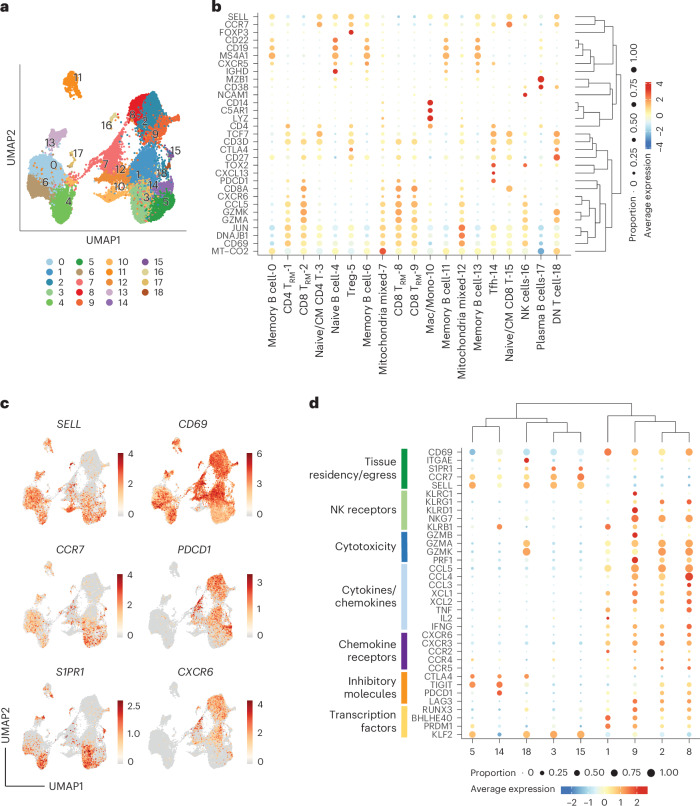
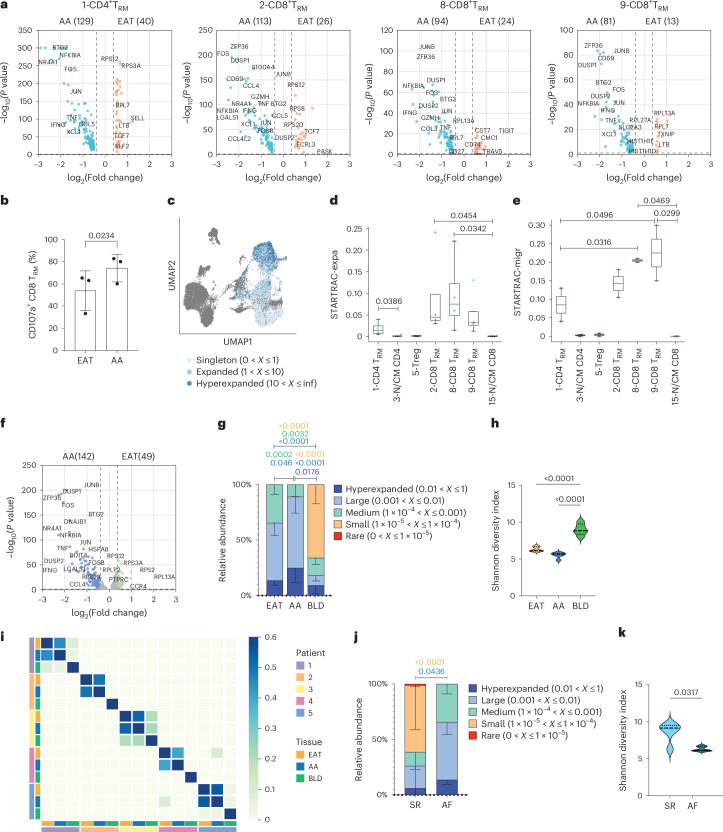
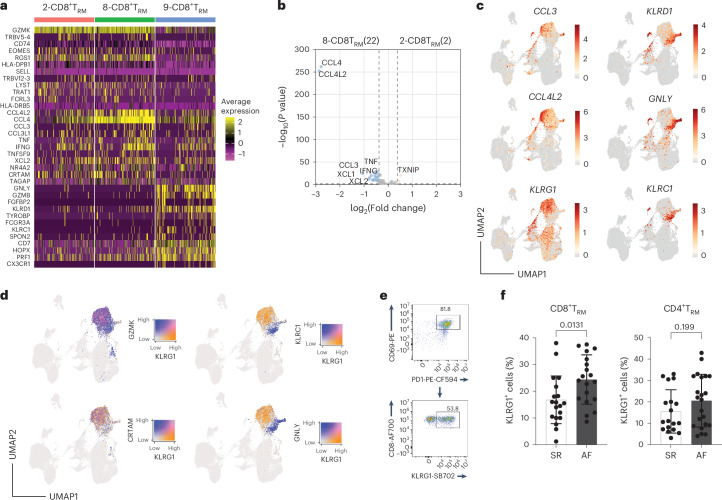
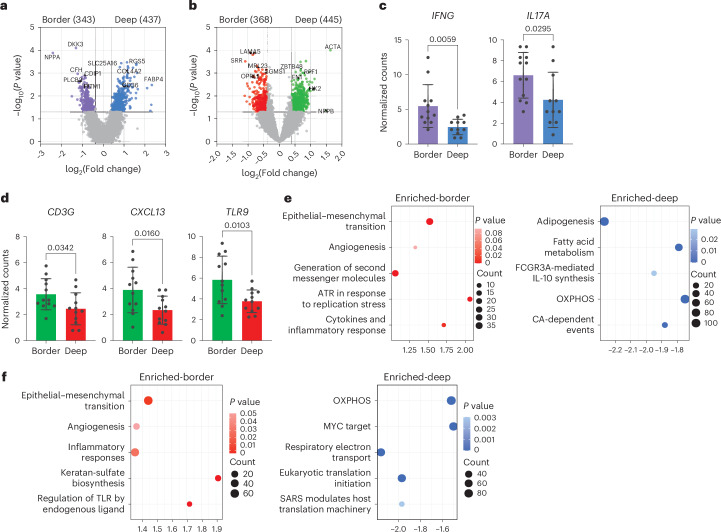
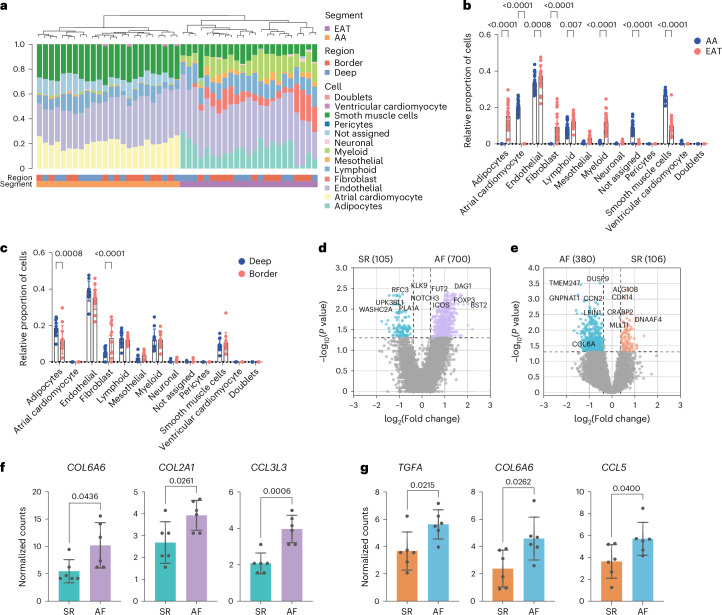
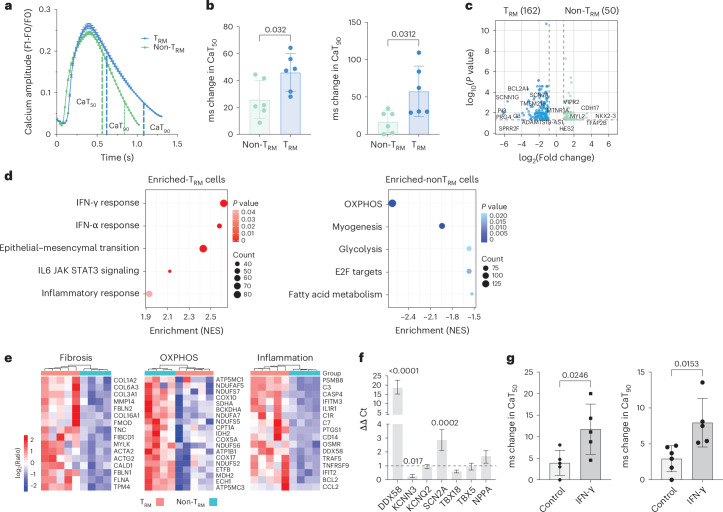
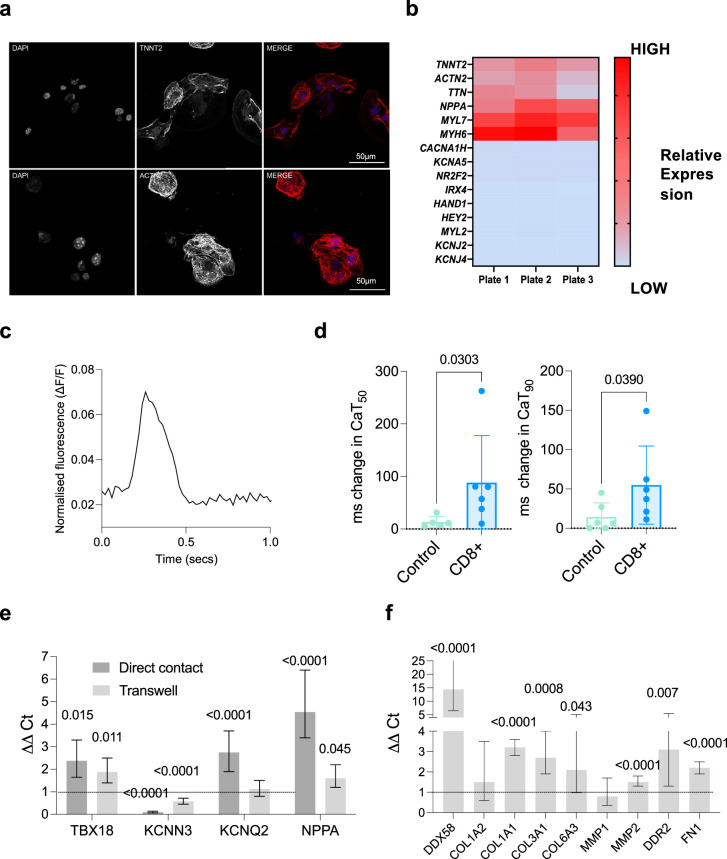
Atrial fibrillation (AF) is the most common sustained arrhythmia and carries an increased risk of stroke and heart failure. Here we investigated how the immune infiltrate of human epicardial adipose tissue (EAT), which directly overlies the myocardium, contributes to AF. Flow cytometry analysis revealed an enrichment of tissue-resident memory T (TRM) cells in patients with AF. Cellular indexing of transcriptomes and epitopes by sequencing (CITE-seq) and single-cell T cell receptor (TCR) sequencing identified two transcriptionally distinct CD8+ TRM cells that are modulated in AF. Spatial transcriptomic analysis of EAT and atrial tissue identified the border region between the tissues to be a region of intense inflammatory and fibrotic activity, and the addition of TRM populations to atrial cardiomyocytes demonstrated their ability to differentially alter calcium flux as well as activate inflammatory and apoptotic signaling pathways. This study identified EAT as a reservoir of TRM cells that can directly modulate vulnerability to cardiac arrhythmia.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures




References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials