

The Wild West of spike-in normalization
- PMID: 39271835
- PMCID: PMC12266361
- DOI: 10.1038/s41587-024-02377-y
The Wild West of spike-in normalization
Abstract
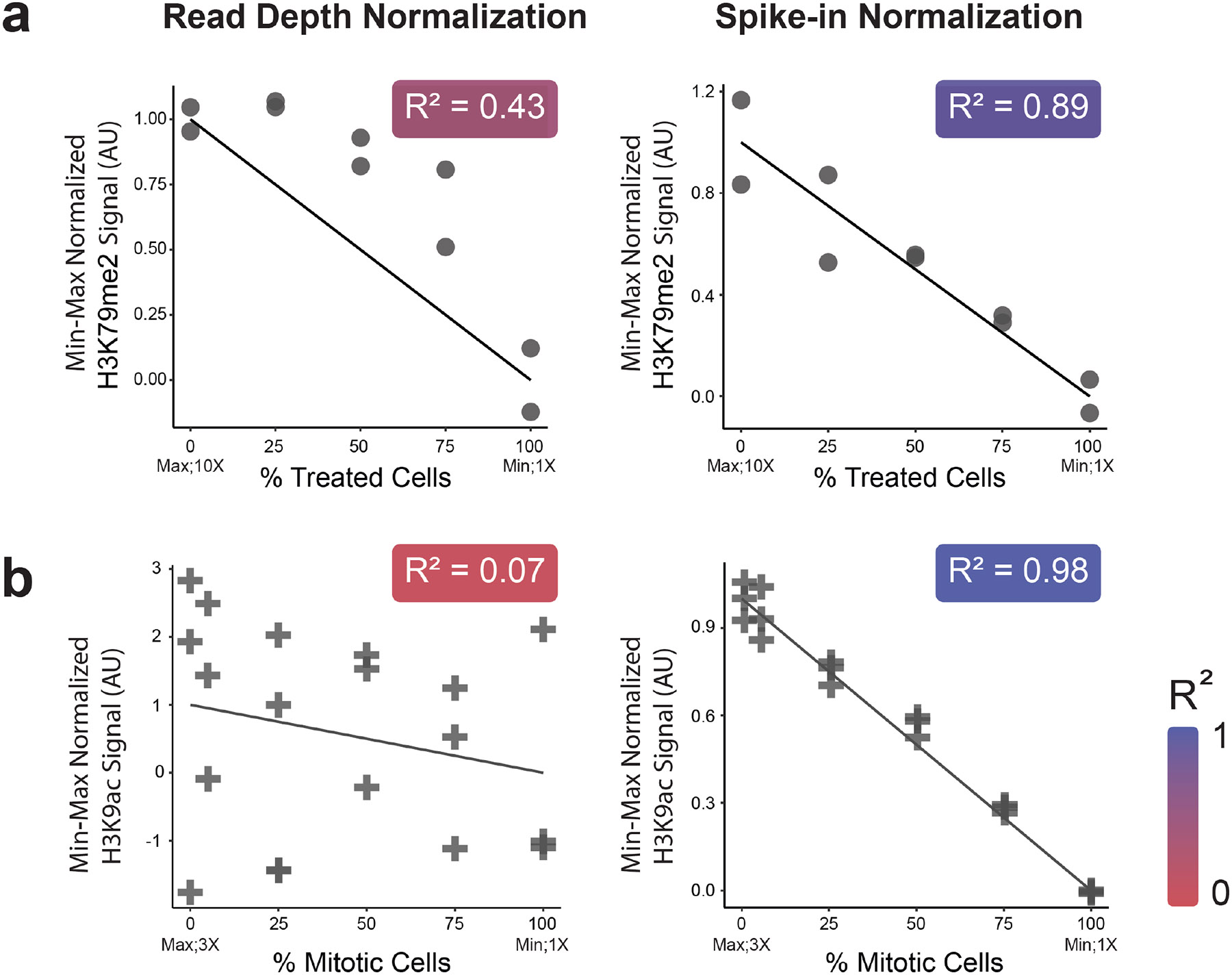
Spike-in normalization is a powerful approach to assess global changes in data obtained from genomic mapping of DNA-associated proteins by methods such as ChIP-sequencing (ChIP-seq), or CUT&RUN. While multiple spike-in methods provide detailed documentation, the implementation of these approaches often omit critical quality control steps and veer from the established procedures. Spike-in normalization typically makes use of a single scalar to normalize genome-wide data, making the approach particularly vulnerable to errors in implementation. Here, we show that proper application of spike-in normalization can increase quantification accuracy across a spectrum of conditions and outline how misuse of spike-in approaches can create erroneous biological interpretations. We conclude by providing guidelines to minimize pitfalls when applying this approach to normalize data from protein-DNA interaction results.
Figures



References
-
- Bonhoure N; Bounova G; Bernasconi D; Praz V; Lammers F; Canella D; Willis IM; Herr W; Hernandez N; Delorenzi M; Hernandez N; Delorenzi M; Deplancke B; Desvergne B; Guex N; Herr W; Naef F; Rougemont J; Schibler U; Andersin T; Cousin P; Gilardi F; Gos P; Lammers F; Raghav S; Villeneuve D; Fabbretti R; Vlegel V; Xenarios I; Migliavacca E; Praz V; David F; Jarosz Y; Kuznetsov D; Liechti R; Martin O; Delafontaine J; Cajan J; Gustafson K; Krier I; Leleu M; Molina N; Naldi A; Rib L; Symul L; Bounova G Quantifying ChIP-Seq Data: A Spiking Method Providing an Internal Reference for Sample-to-Sample Normalization. Genome Res 2014, 24 (7), 1157–1168. 10.1101/gr.168260.113. - DOI - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
