

Genomic and phenotypic landscapes of X-linked hereditary hearing loss in the Chinese population
- PMID: 39272213
- PMCID: PMC11396341
- DOI: 10.1186/s13023-024-03338-z
Genomic and phenotypic landscapes of X-linked hereditary hearing loss in the Chinese population
Abstract
Background: Hearing loss (HL) is the most common sensory birth deficit worldwide, with causative variants in more than 150 genes. However, the etiological contribution and clinical manifestations of X-linked inheritance in HL remain unclear within the Chinese HL population. In this study, we focused on X-linked hereditary HL and aimed to assess its contribution to hereditary HL and identify the genotype-phenotype relationship.
Methods: We performed a molecular epidemiological investigation of X-linked hereditary HL based on next-generation sequencing and third-generation sequencing in 3646 unrelated patients with HL. We also discussed the clinical features associated with X-linked non-syndromic HL-related genes based on a review of the literature.
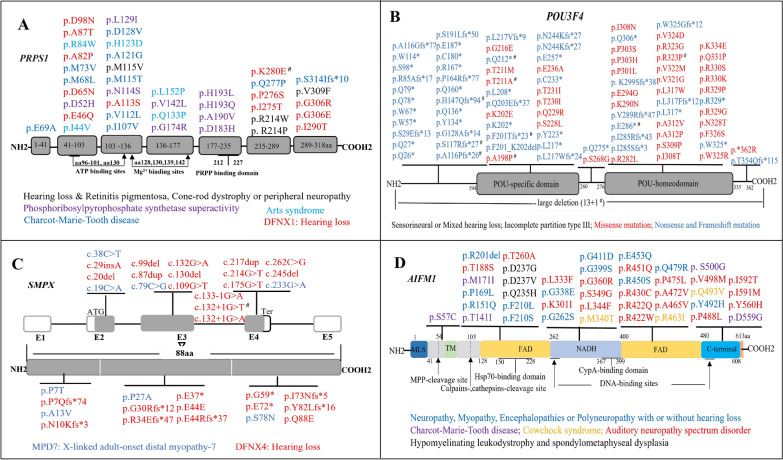
Results: We obtained a diagnostic rate of 52.72% (1922/3646) among our patients; the aggregate contribution of HL caused by genes on the X chromosome in this cohort was ~ 1.14% (22/1922), and POU3F4 variants caused ~ 59% (13/22) of these cases. We found that X-linked HL was congenital or began during childhood in all cases, with representative audiological profiles or typical cochlear malformations in certain genes. Genotypic and phenotypic analyses showed that causative variants in PRPS1 and AIFM1 were mainly of the missense type, suggesting that phenotypic variability was correlated with the different effects that the replaced residues exert on structure and function. Variations in SMPX causing truncation of the protein product were associated with DFNX4, which resulted in typical audiological profiles before and after the age of 10 years, whereas nontruncated proteins typically led to distal myopathy. No phenotypic differences were identified in patients carrying POU3F4 or COL4A6 variants.
Conclusions: Our work constitutes a preliminary evaluation of the molecular contribution of X-linked genes in heritable HL (~ 1.14%). The 15 novel variants reported here expand the mutational spectrum of these genes. Analysis of the genotype-phenotype relationship is valuable for X-linked HL precise diagnostics and genetic counseling. Elucidation of the pathogenic mechanisms and audiological profiles of HL can also guide choices regarding treatment modalities.
Keywords: DFNX; Genetic variants spectrum; Genotype–phenotype correlations; X-linked hearing loss.
© 2024. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures


Similar articles
-
Clinical and molecular characterization of POU3F4 mutations in multiple DFNX2 Chinese families.BMC Med Genet. 2018 Sep 4;19(1):157. doi: 10.1186/s12881-018-0630-9. BMC Med Genet. 2018. PMID: 30176854 Free PMC article.
-
X-linked hearing loss: two gene mutation examples provide generalizable implications for clinical care.Am J Audiol. 2014 Jun;23(2):190-200. doi: 10.1044/2014_AJA-13-0040. Am J Audiol. 2014. PMID: 24687041
-
Novel and De Novo Mutations Extend Association of POU3F4 with Distinct Clinical and Radiological Phenotype of Hearing Loss.PLoS One. 2016 Dec 12;11(12):e0166618. doi: 10.1371/journal.pone.0166618. eCollection 2016. PLoS One. 2016. PMID: 27941975 Free PMC article.
-
X-Linked Sensorineural Hearing Loss: A Literature Review.Curr Genomics. 2018 Aug;19(5):327-338. doi: 10.2174/1389202919666171218163046. Curr Genomics. 2018. PMID: 30065609 Free PMC article. Review.
-
Genetic etiology of hereditary hearing loss in the Gulf Cooperation Council countries.Hum Genet. 2022 Apr;141(3-4):595-605. doi: 10.1007/s00439-021-02323-x. Epub 2021 Aug 2. Hum Genet. 2022. PMID: 34338889 Review.
References
-
- Schraders M, Haas SA, Weegerink NJ, Oostrik J, Hu H, Hoefsloot LH, Kannan S, Huygen PL, Pennings RJ, Admiraal RJ, et al. Next-generation sequencing identifies mutations of SMPX, which encodes the small muscle protein, X-linked, as a cause of progressive hearing impairment. Am J Hum Genet. 2011;88(5):628–34. 10.1016/j.ajhg.2011.04.012 - DOI - PMC - PubMed
-
- Huebner AK, Gandia M, Frommolt P, Maak A, Wicklein EM, Thiele H, Altmüller J, Wagner F, Viñuela A, Aguirre LA, et al. Nonsense mutations in SMPX, encoding a protein responsive to physical force, result in X-chromosomal hearing loss. Am J Hum Genet. 2011;88(5):621–7. 10.1016/j.ajhg.2011.04.007 - DOI - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
