

Pathomechanisms of Monoallelic variants in TTN causing skeletal muscle disease
- PMID: 39277846
- PMCID: PMC11578113
- DOI: 10.1093/hmg/ddae136
Pathomechanisms of Monoallelic variants in TTN causing skeletal muscle disease
Erratum in
-
Correction to: Pathomechanisms of Monoallelic variants in TTN causing skeletal muscle disease.Hum Mol Genet. 2025 Jan 29;34(2):205. doi: 10.1093/hmg/ddae181. Hum Mol Genet. 2025. PMID: 39656636 Free PMC article. No abstract available.
Abstract
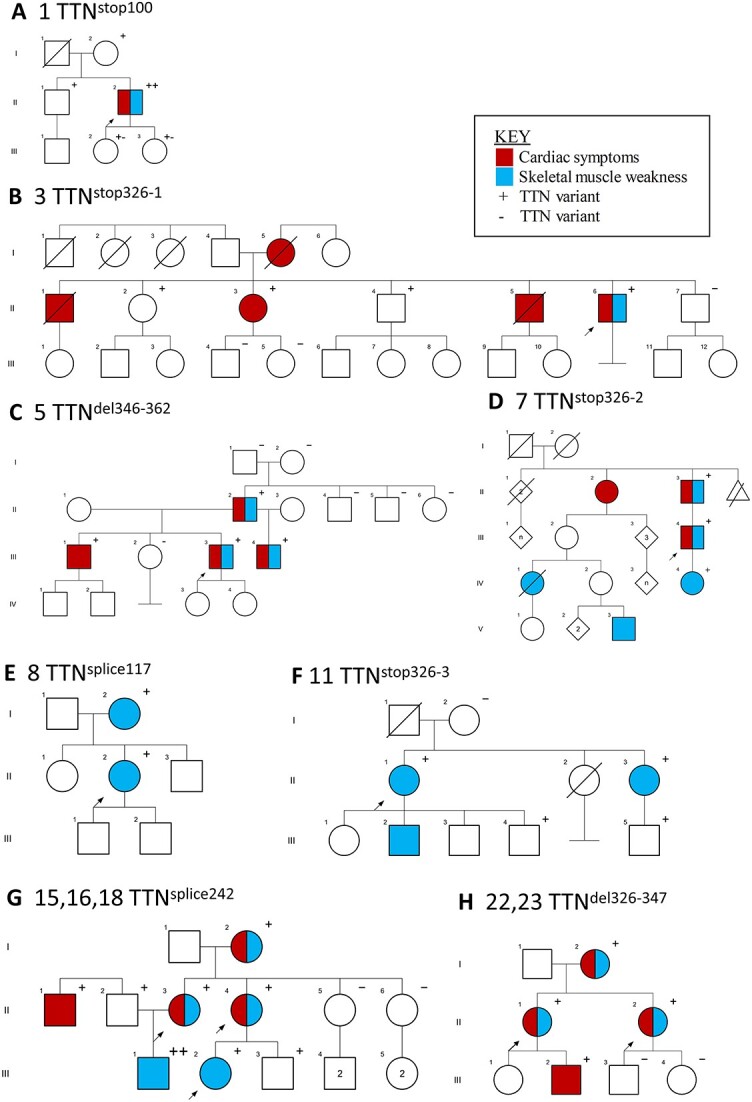
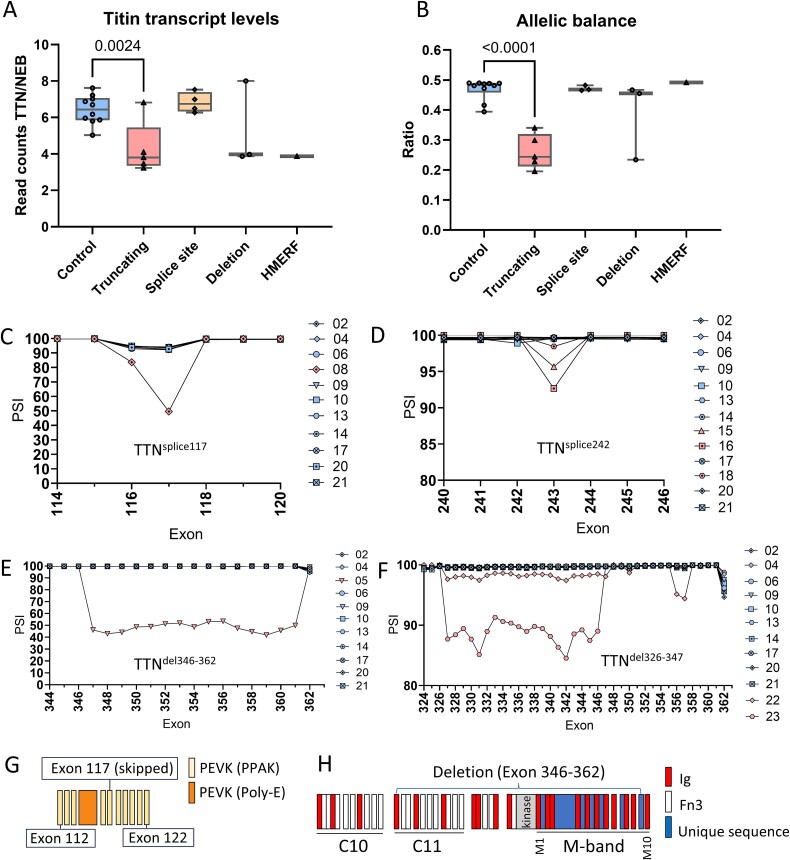
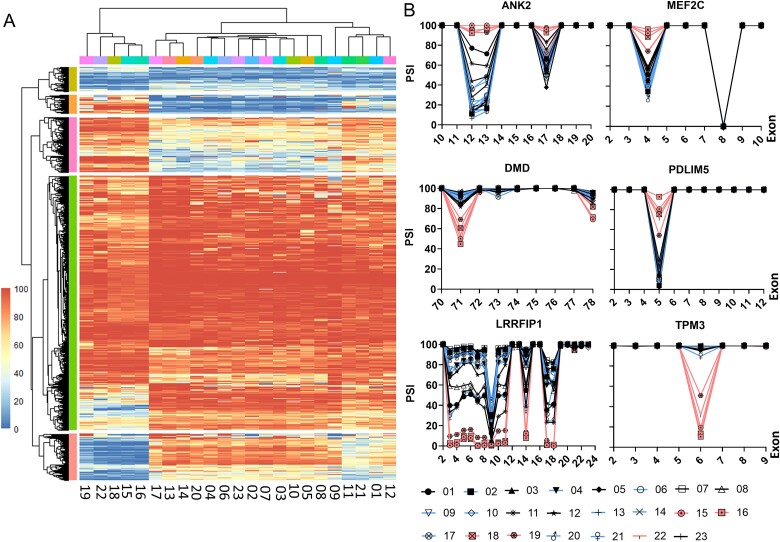
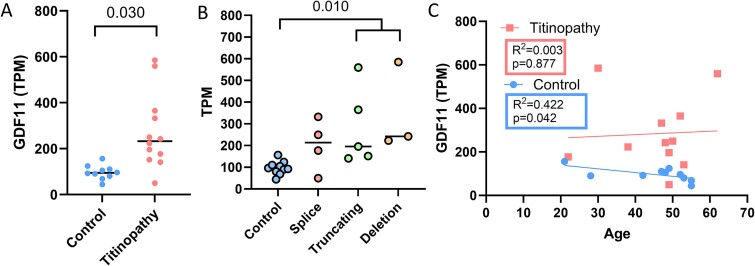
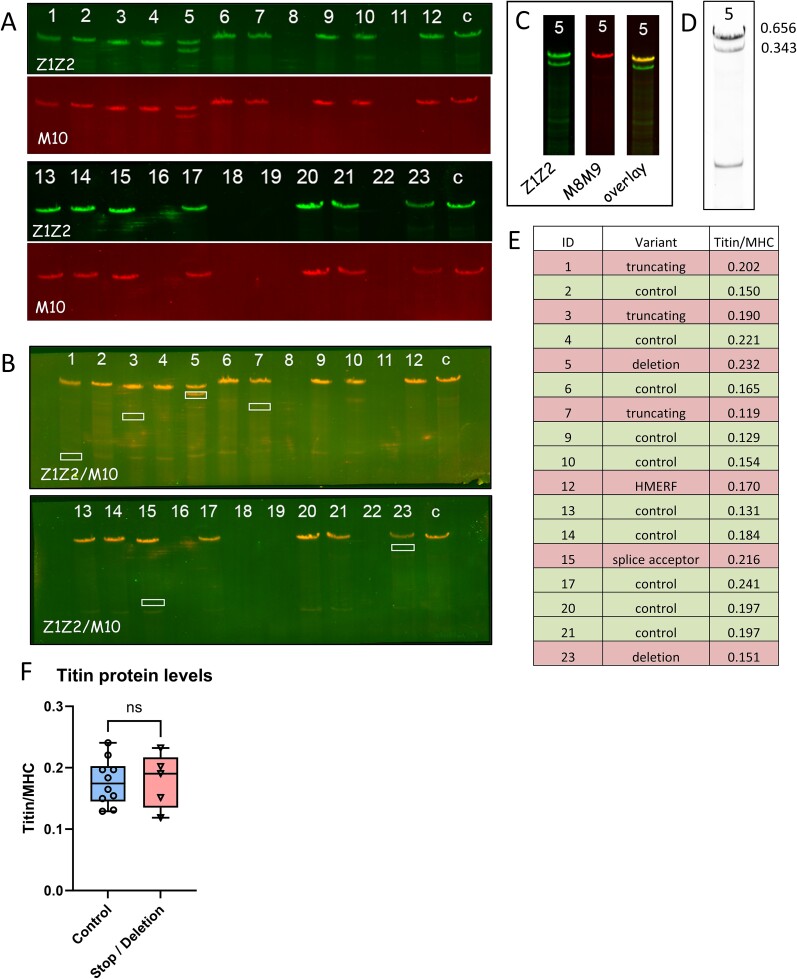
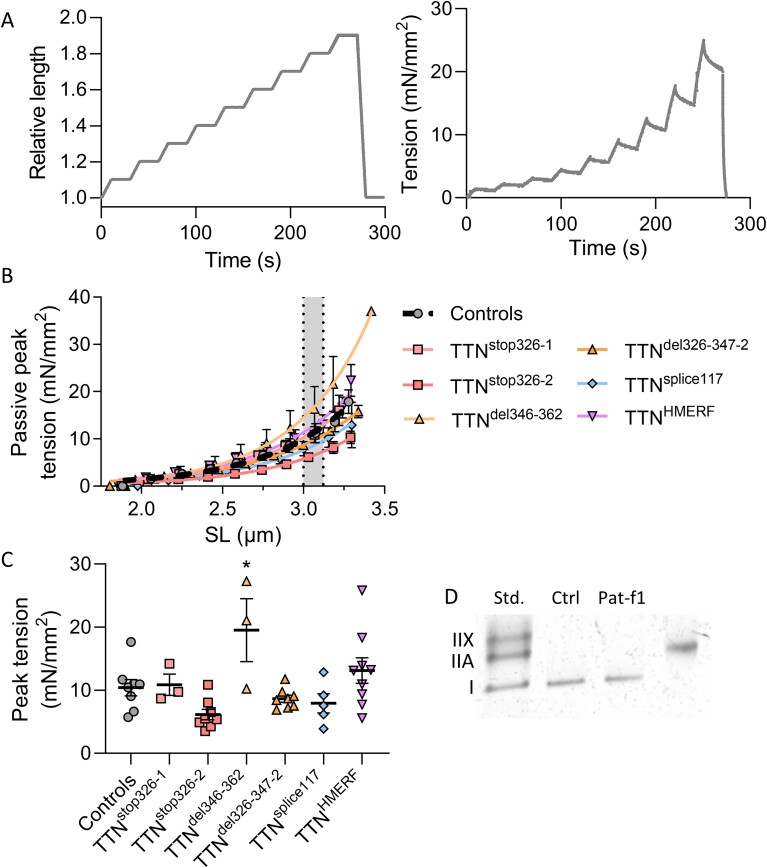
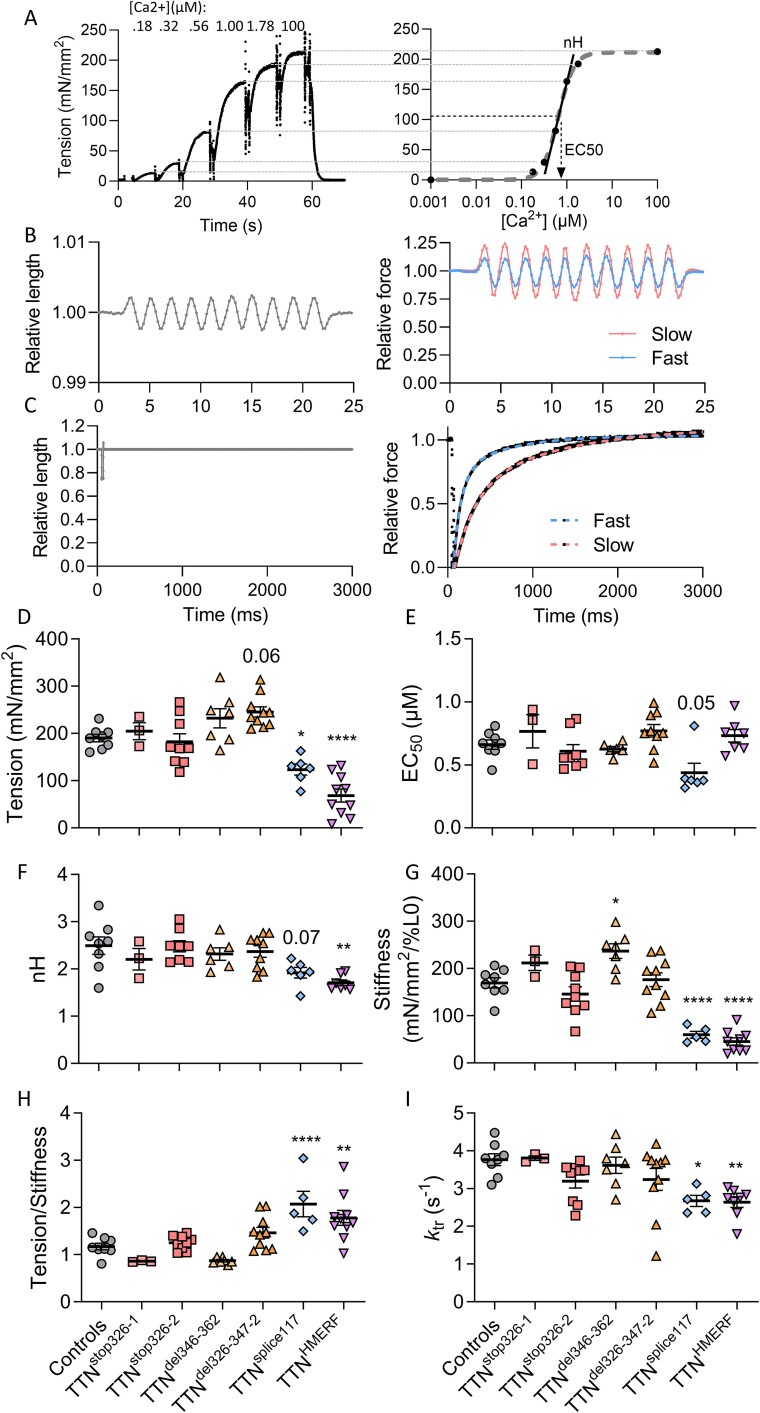
Pathogenic variants in the titin gene (TTN) are known to cause a wide range of cardiac and musculoskeletal disorders, with skeletal myopathy mostly attributed to biallelic variants. We identified monoallelic truncating variants (TTNtv), splice site or internal deletions in TTN in probands with mild, progressive axial and proximal weakness, with dilated cardiomyopathy frequently developing with age. These variants segregated in an autosomal dominant pattern in 7 out of 8 studied families. We investigated the impact of these variants on mRNA, protein levels, and skeletal muscle structure and function. Results reveal that nonsense-mediated decay likely prevents accumulation of harmful truncated protein in skeletal muscle in patients with TTNtvs. Splice variants and an out-of-frame deletion induce aberrant exon skipping, while an in-frame deletion produces shortened titin with intact N- and C-termini, resulting in disrupted sarcomeric structure. All variant types were associated with genome-wide changes in splicing patterns, which represent a hallmark of disease progression. Lastly, RNA-seq studies revealed that GDF11, a member of the TGF-β superfamily, is upregulated in diseased tissue, indicating that it might be a useful therapeutic target in skeletal muscle titinopathies.
Keywords: TTN deletion; TTNtv; Dominant titinopathy; Titin; titinopathy pathomechanisms.
© The Author(s) 2024. Published by Oxford University Press.
Figures

References
-
- Granzier H, Wu Y, Siegfried L. et al. Titin: physiological function and role in cardiomyopathy and failure. Heart Fail Rev 2005;10:211–223. - PubMed
-
- Bang ML, Centner T, Fornoff F. et al. The complete gene sequence of titin, expression of an unusual approximately 700-kDa titin isoform, and its interaction with obscurin identify a novel Z-line to I-band linking system. Circ Res 2001;89:1065–1072. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
