

Metabolic ripple effects - deciphering how lipid metabolism in cancer interfaces with the tumor microenvironment
- PMID: 39284708
- PMCID: PMC11423921
- DOI: 10.1242/dmm.050814
Metabolic ripple effects - deciphering how lipid metabolism in cancer interfaces with the tumor microenvironment
Abstract
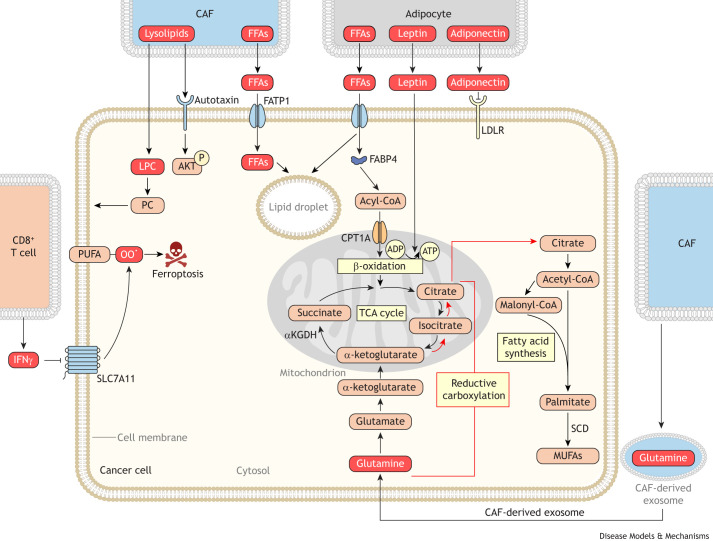
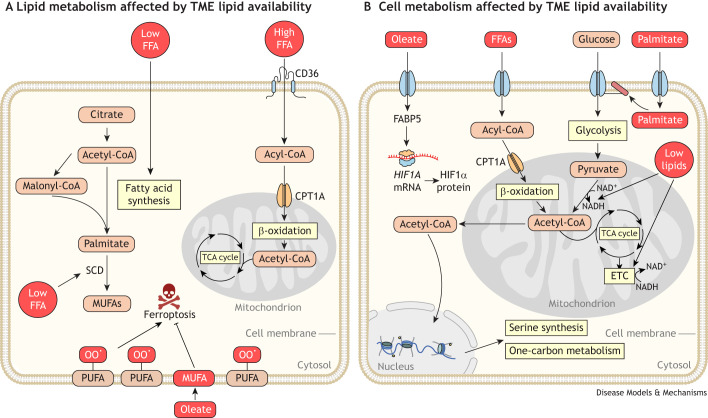
Cancer cells require a constant supply of lipids. Lipids are a diverse class of hydrophobic molecules that are essential for cellular homeostasis, growth and survival, and energy production. How tumors acquire lipids is under intensive investigation, as these mechanisms could provide attractive therapeutic targets for cancer. Cellular lipid metabolism is tightly regulated and responsive to environmental stimuli. Thus, lipid metabolism in cancer is heavily influenced by the tumor microenvironment. In this Review, we outline the mechanisms by which the tumor microenvironment determines the metabolic pathways used by tumors to acquire lipids. We also discuss emerging literature that reveals that lipid availability in the tumor microenvironment influences many metabolic pathways in cancers, including those not traditionally associated with lipid biology. Thus, metabolic changes instigated by the tumor microenvironment have 'ripple' effects throughout the densely interconnected metabolic network of cancer cells. Given the interconnectedness of tumor metabolism, we also discuss new tools and approaches to identify the lipid metabolic requirements of cancer cells in the tumor microenvironment and characterize how these requirements influence other aspects of tumor metabolism.
Keywords: Acidosis; Diet; Hypoxia; Lipid metabolism; Nutrient deprivation; Tumor microenvironment.
© 2024. Published by The Company of Biologists Ltd.
Conflict of interest statement
Competing interests The authors declare no competing or financial interests.
Figures
Similar articles
-
Lipid metabolism in cancer: New perspectives and emerging mechanisms.Dev Cell. 2021 May 17;56(10):1363-1393. doi: 10.1016/j.devcel.2021.04.013. Epub 2021 May 3. Dev Cell. 2021. PMID: 33945792 Review.
-
Adaptation mechanisms in cancer: Lipid metabolism under hypoxia and nutrient deprivation as a target for novel therapeutic strategies (Review).Mol Med Rep. 2025 Apr;31(4):83. doi: 10.3892/mmr.2025.13448. Epub 2025 Jan 31. Mol Med Rep. 2025. PMID: 39886950 Free PMC article. Review.
-
Lipids in the tumor microenvironment: From cancer progression to treatment.Prog Lipid Res. 2020 Nov;80:101055. doi: 10.1016/j.plipres.2020.101055. Epub 2020 Aug 11. Prog Lipid Res. 2020. PMID: 32791170 Free PMC article. Review.
-
Hypoxia, lipids, and cancer: surviving the harsh tumor microenvironment.Trends Cell Biol. 2014 Aug;24(8):472-8. doi: 10.1016/j.tcb.2014.06.001. Epub 2014 Jul 4. Trends Cell Biol. 2014. PMID: 24985940 Free PMC article. Review.
-
Lipid metabolic reprogramming in tumor microenvironment: from mechanisms to therapeutics.J Hematol Oncol. 2023 Sep 12;16(1):103. doi: 10.1186/s13045-023-01498-2. J Hematol Oncol. 2023. PMID: 37700339 Free PMC article. Review.
Cited by
-
Microenvironmental arginine restriction sensitizes pancreatic cancers to polyunsaturated fatty acids by suppression of lipid synthesis.bioRxiv [Preprint]. 2025 Mar 13:2025.03.10.642426. doi: 10.1101/2025.03.10.642426. bioRxiv. 2025. PMID: 40161789 Free PMC article. Preprint.
-
Protein-based Radiopharmaceuticals that target fibroblast activation protein alpha: a review of current progress.EJNMMI Radiopharm Chem. 2025 Jun 21;10(1):32. doi: 10.1186/s41181-025-00356-5. EJNMMI Radiopharm Chem. 2025. PMID: 40542914 Free PMC article. Review.
-
Targeting piRNA-137463 Inhibits Tumor Progression and Boosts Sensitivity to Immune Checkpoint Blockade via De Novo Cholesterol Biosynthesis in Lung Adenocarcinoma.Adv Sci (Weinh). 2025 Feb;12(6):e2414100. doi: 10.1002/advs.202414100. Epub 2024 Dec 18. Adv Sci (Weinh). 2025. PMID: 39692168 Free PMC article.
-
Cancer-associated Fibroblast-like Cells Promote Osteosarcoma Metastasis by Upregulation of Phosphoserine Aminotransferase 1 and Activation of the mTOR/S6K Pathway.Int J Biol Sci. 2025 Jun 20;21(9):4153-4171. doi: 10.7150/ijbs.109169. eCollection 2025. Int J Biol Sci. 2025. PMID: 40612679 Free PMC article.
References
-
- Ackerman, D., Tumanov, S., Qiu, B., Michalopoulou, E., Spata, M., Azzam, A., Xie, H., Simon, M. C. and Kamphorst, J. J. (2018). Triglycerides promote lipid homeostasis during hypoxic stress by balancing fatty acid saturation. Cell Rep 24, 2596-2605.e5. 10.1016/j.celrep.2018.08.015 - DOI - PMC - PubMed
-
- Agarwala, P. K., Nie, S., Reid, G. E. and Kapoor, S. (2023). Global lipid remodelling by hypoxia aggravates migratory potential in pancreatic cancer while maintaining plasma membrane homeostasis. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1868, 159398. 10.1016/j.bbalip.2023.159398 - DOI - PMC - PubMed
-
- Alicea, G. M., Rebecca, V. W., Goldman, A. R., Fane, M. E., Douglass, S. M., Behera, R., Webster, M. R., Kugel, C. H., Ecker, B. L., Cecilia Caino, M.et al. (2020). Changes in aged fibroblast lipid metabolism induce age-dependent melanoma cell resistance to targeted therapy via the fatty acid transporter FATP2. Cancer Discov. 10, 1282-1295. 10.1158/2159-8290.CD-20-0329 - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
