

Systematic prioritization of functional variants and effector genes underlying colorectal cancer risk
- PMID: 39284974
- PMCID: PMC11525171
- DOI: 10.1038/s41588-024-01900-w
Systematic prioritization of functional variants and effector genes underlying colorectal cancer risk
Abstract
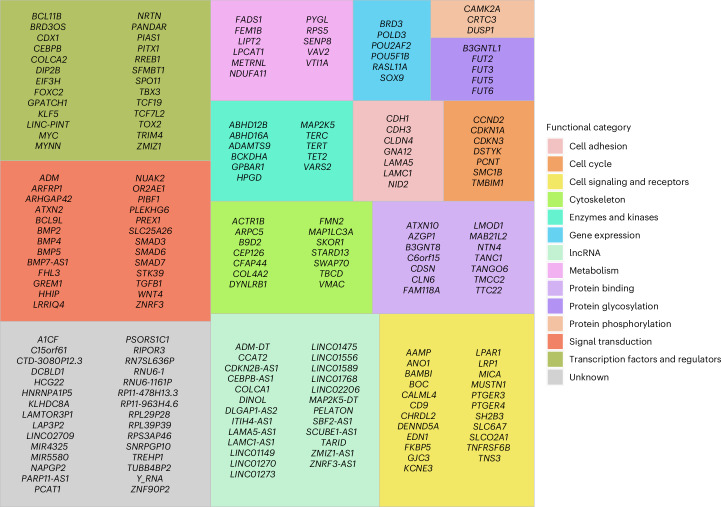
Genome-wide association studies of colorectal cancer (CRC) have identified 170 autosomal risk loci. However, for most of these, the functional variants and their target genes are unknown. Here, we perform statistical fine-mapping incorporating tissue-specific epigenetic annotations and massively parallel reporter assays to systematically prioritize functional variants for each CRC risk locus. We identify plausible causal variants for the 170 risk loci, with a single variant for 40. We link these variants to 208 target genes by analyzing colon-specific quantitative trait loci and implementing the activity-by-contact model, which integrates epigenomic features and Micro-C data, to predict enhancer-gene connections. By deciphering CRC risk loci, we identify direct links between risk variants and target genes, providing further insight into the molecular basis of CRC susceptibility and highlighting potential pharmaceutical targets for prevention and treatment.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures







References
-
- Lichtenstein, P. et al. Environmental and heritable factors in the causation of cancer–analyses of cohorts of twins from Sweden, Denmark, and Finland. N. Engl. J. Med.343, 78–85 (2000). - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
