

Post-transcriptional reprogramming by thousands of mRNA untranslated regions in trypanosomes
- PMID: 39285175
- PMCID: PMC11405848
- DOI: 10.1038/s41467-024-52432-0
Post-transcriptional reprogramming by thousands of mRNA untranslated regions in trypanosomes
Abstract
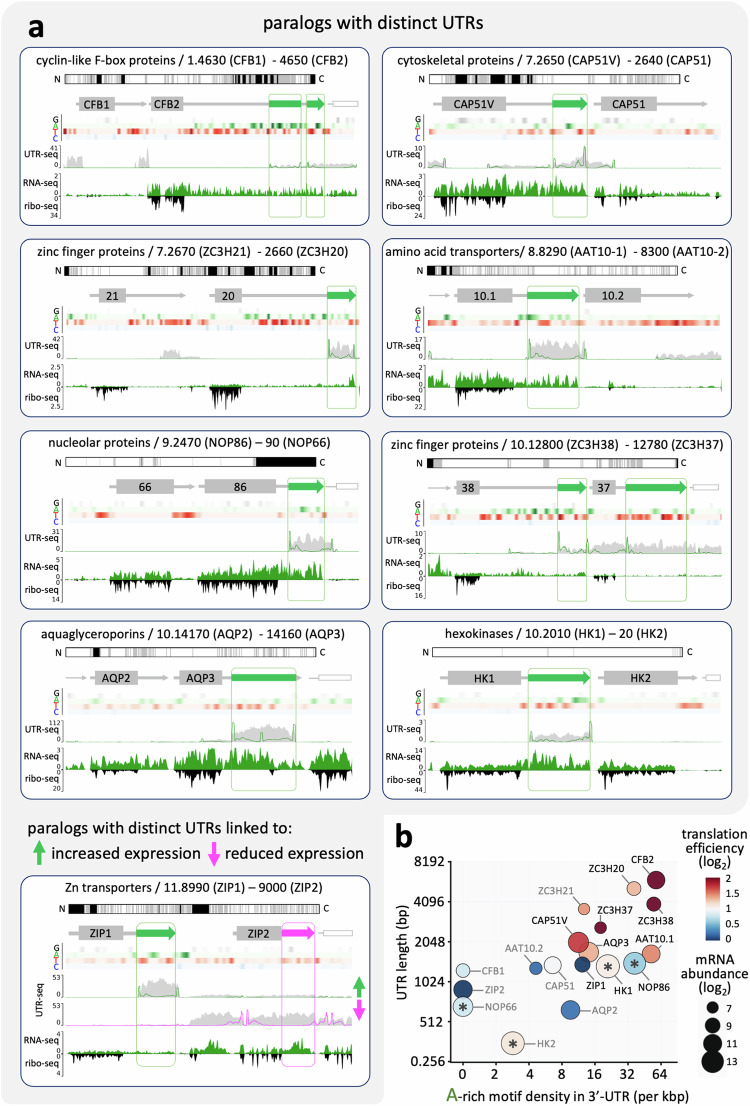
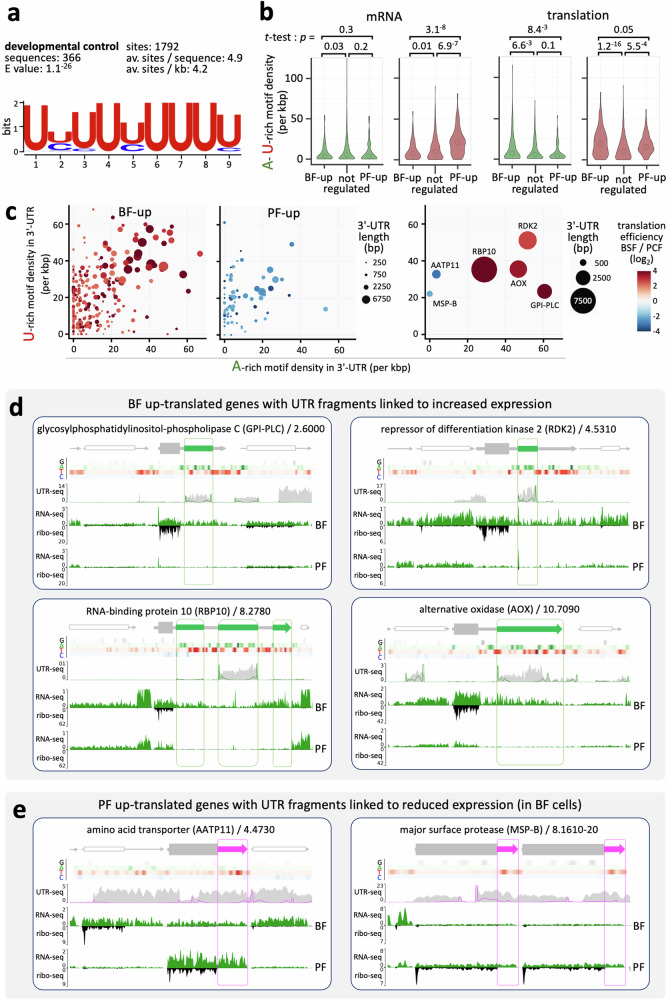
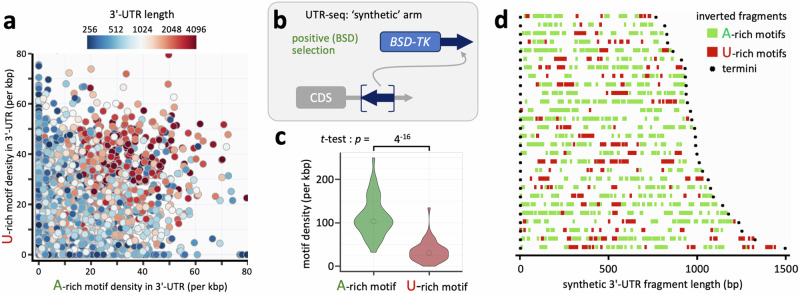
Although genome-wide polycistronic transcription places major emphasis on post-transcriptional controls in trypanosomatids, messenger RNA cis-regulatory untranslated regions (UTRs) have remained largely uncharacterised. Here, we describe a genome-scale massive parallel reporter assay coupled with 3'-UTR-seq profiling in the African trypanosome and identify thousands of regulatory UTRs. Increased translation efficiency was associated with dosage of adenine-rich poly-purine tracts (pPuTs). An independent assessment of native UTRs using machine learning based predictions confirmed the robust correspondence between pPuTs and positive control, as did an assessment of synthetic UTRs. Those 3'-UTRs associated with upregulated expression in bloodstream-stage cells were also enriched in uracil-rich poly-pyrimidine tracts, suggesting a mechanism for developmental activation through pPuT 'unmasking'. Thus, we describe a cis-regulatory UTR sequence 'code' that underpins gene expression control in the context of a constitutively transcribed genome. We conclude that thousands of UTRs post-transcriptionally reprogram gene expression profiles in trypanosomes.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures






References
Publication types
MeSH terms
Substances
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
