

Agonist antibody to MuSK protects mice from MuSK myasthenia gravis
- PMID: 39288173
- PMCID: PMC11441477
- DOI: 10.1073/pnas.2408324121
Agonist antibody to MuSK protects mice from MuSK myasthenia gravis
Abstract
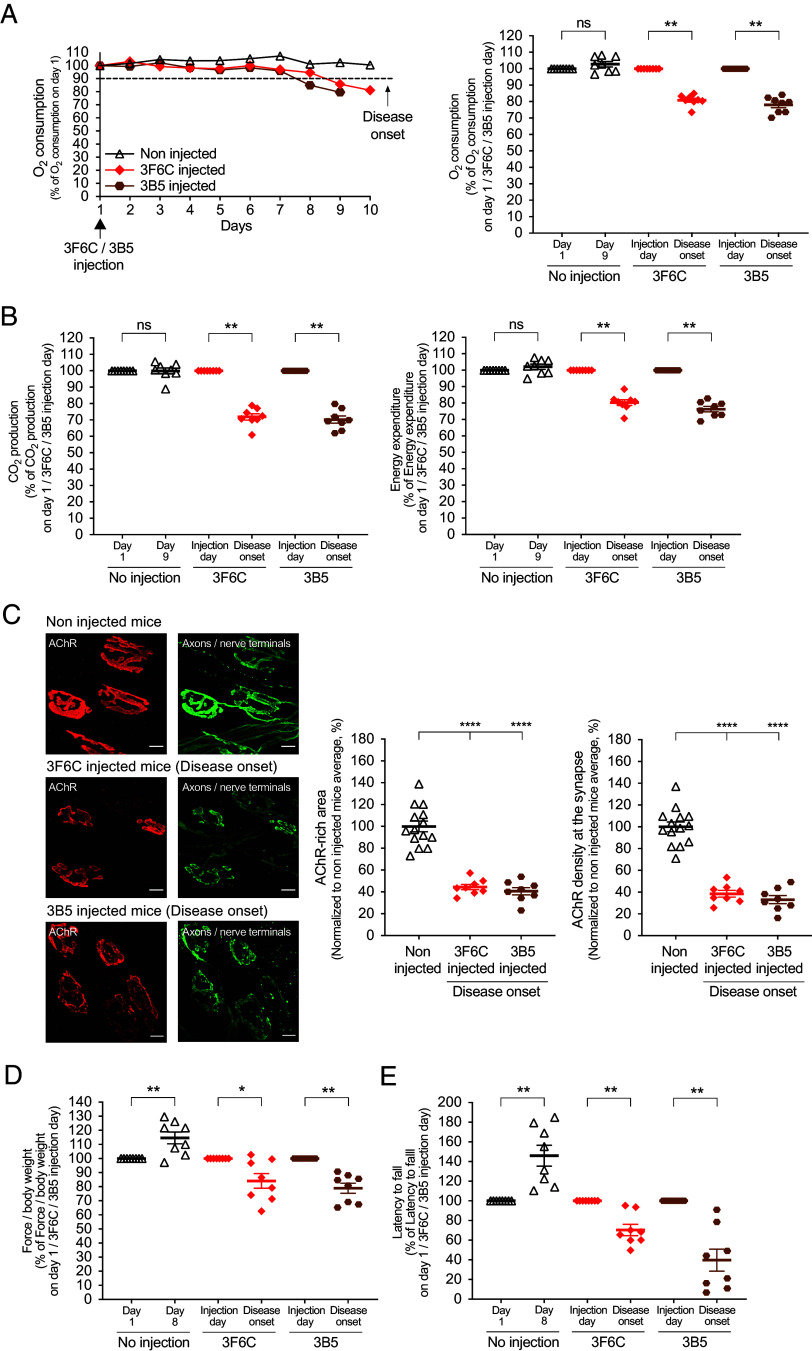
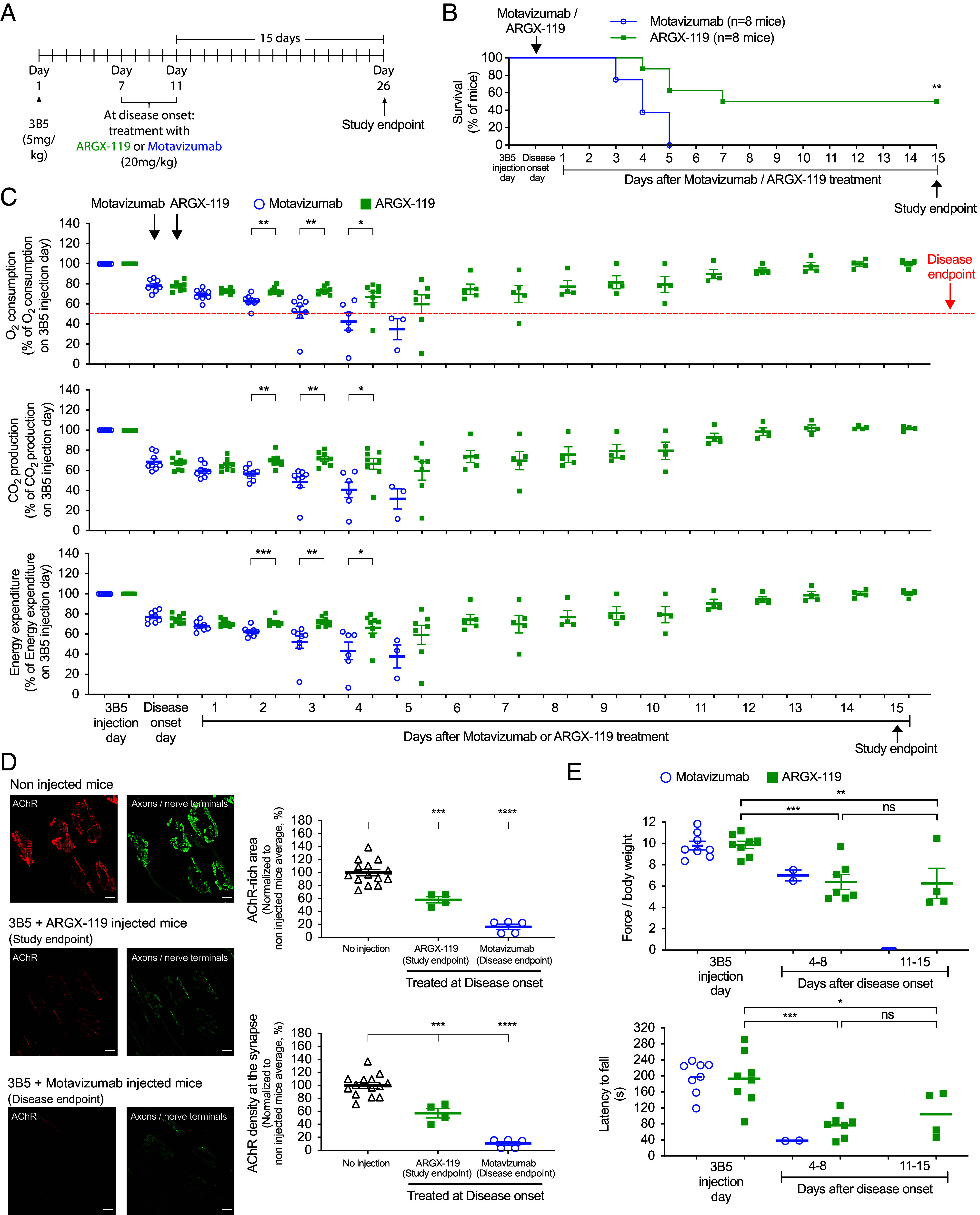
Myasthenia gravis (MG) is a chronic and severe disease of the skeletal neuromuscular junction (NMJ) in which the effects of neurotransmitters are attenuated, leading to muscle weakness. In the most common forms of autoimmune MG, antibodies attack components of the postsynaptic membrane, including the acetylcholine receptor (AChR) or muscle-specific kinase (MuSK). MuSK, a master regulator of NMJ development, associates with the low-density lipoprotein-related receptor 4 (Lrp4) to form the signaling receptor for neuronal Agrin, a nerve-derived synaptic organizer. Pathogenic antibodies to MuSK interfere with binding between MuSK and Lrp4, inhibiting the differentiation and maintenance of the NMJ. MuSK MG can be debilitating and refractory to treatments that are effective for AChR MG. We show here that recombinant antibodies, derived from MuSK MG patients, cause severe neuromuscular disease in mice. The disease can be prevented by a MuSK agonist antibody, presented either prophylactically or after disease onset. These findings suggest a therapeutic alternative to generalized immunosuppression for treating MuSK MG by selectively and directly targeting the disease mechanism.
Keywords: autoimmune disease; myasthenia gravis; neuromuscular; synapse; therapeutic antibody.
Conflict of interest statement
Competing interests statement:C.S., B.V., K.S., and R.V. are employees of and have equity ownership in argenx BV. Issued patents: S.J.B., NYU Medical School, US9329182 S.J.B., Wei Zhang, Maartje Huijbers, Johannes J. Verschuuren, and Silvere M. van der Maarel; NYU Medical School and LUMC; US20150125442A1 S.J.B. et al., NYU Medical School and argenx; US11492401 Patent applications: M.G.H. et al., LUMC, WO2020/055241 M.G.H. et al., LUMC and argenx, WO2021/180676 R.V. et al.; argenx, Université de Montréal and NYU Medical School; WO2023/147489 R.V. et al.; argenx and NYU Medical School; WO2023/218099. S.J.B. is grateful for financial support for research from argenx.
Figures




References
-
- Burden S. J., The formation of neuromuscular synapses. Genes Dev. 12, 133–148 (1998). - PubMed
-
- Tintignac L. A., Brenner H.-R., Rüegg M. A., Mechanisms regulating neuromuscular junction development and function and causes of muscle wasting. Physiol. Rev. 95, 809–852 (2015). - PubMed
-
- Sanes J. R., Lichtman J. W., Induction, assembly, maturation and maintenance of a postsynaptic apparatus. Nat. Rev. Neurosci. 2, 791–805 (2001). - PubMed
-
- DeChiara T. M., et al. , The receptor tyrosine kinase MuSK is required for neuromuscular junction formation in vivo. Cell 85, 501–512 (1996). - PubMed
MeSH terms
Substances
Grants and funding
- R37 NS036193/NS/NINDS NIH HHS/United States
- R37 NS36193/HHS | NIH | National Institute of Neurological Disorders and Stroke (NINDS)
- 1R01AG051490/HHS | NIH | National Institute of Neurological Disorders and Stroke (NINDS)
- W.OR-19.13/Prinses Beatrix Spierfonds (Het Prinses Beatrix Spierfonds)
- no number/Argenx (Argenx SE)
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
