

Atomistic mechanisms of the regulation of small-conductance Ca2+-activated K+ channel (SK2) by PIP2
- PMID: 39288178
- PMCID: PMC11441529
- DOI: 10.1073/pnas.2318900121
Atomistic mechanisms of the regulation of small-conductance Ca2+-activated K+ channel (SK2) by PIP2
Abstract
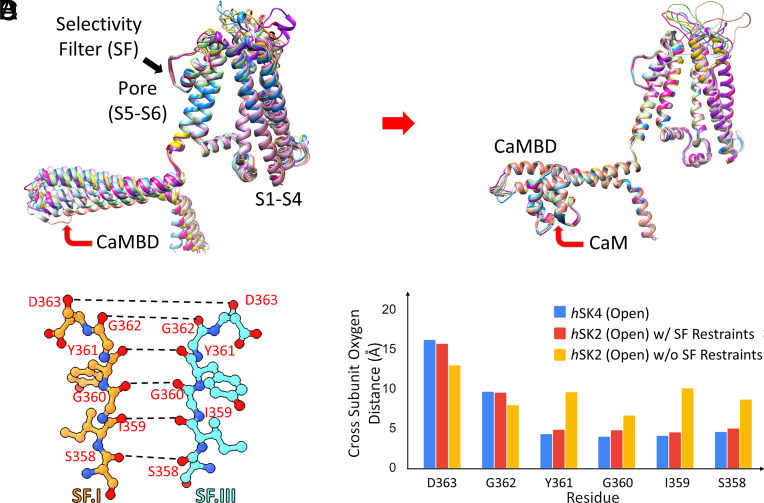
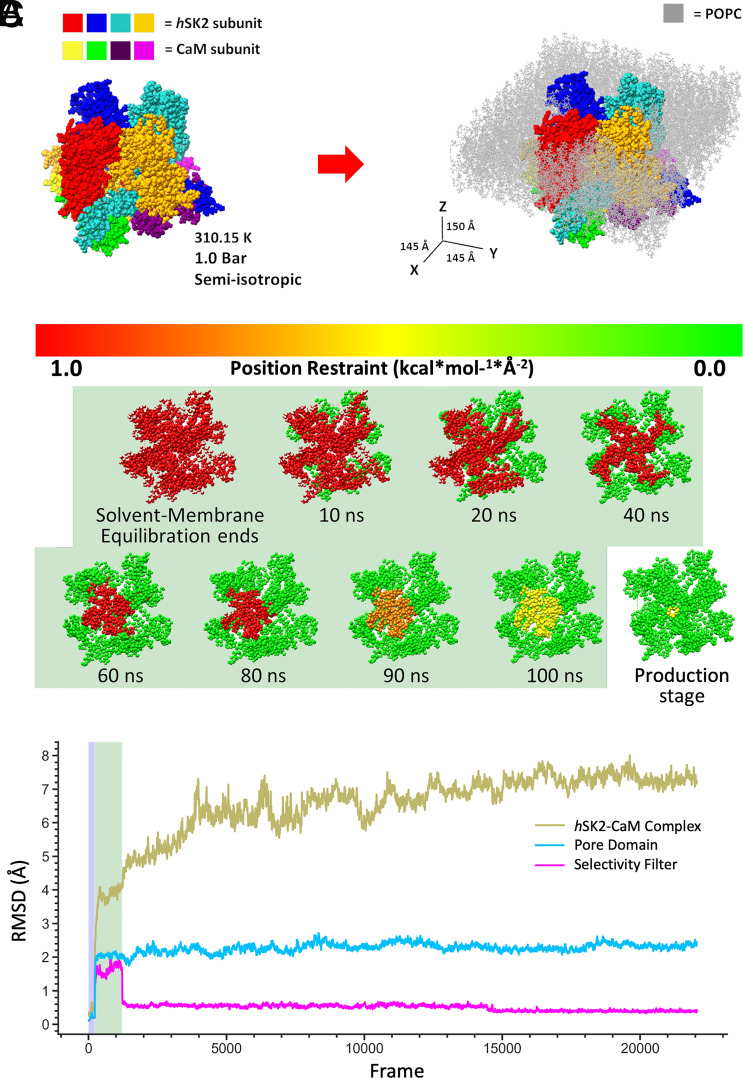
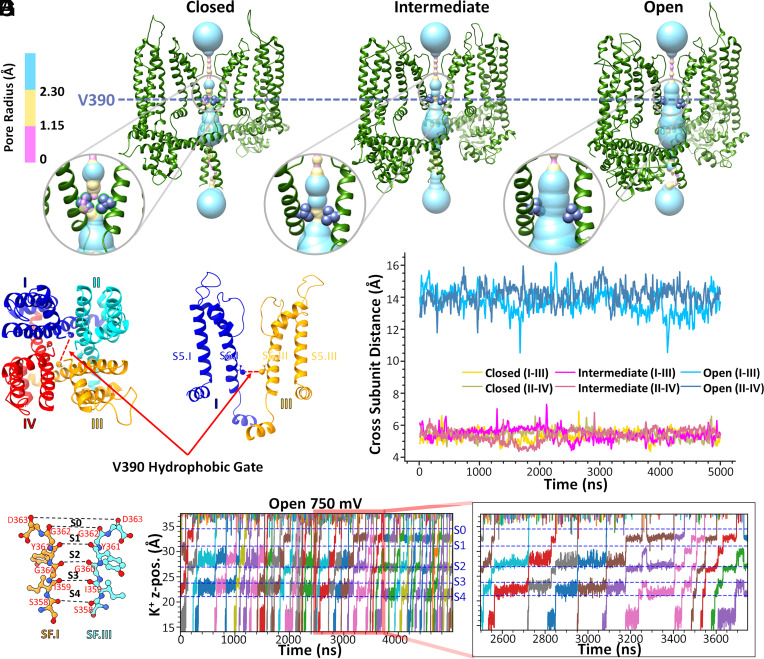
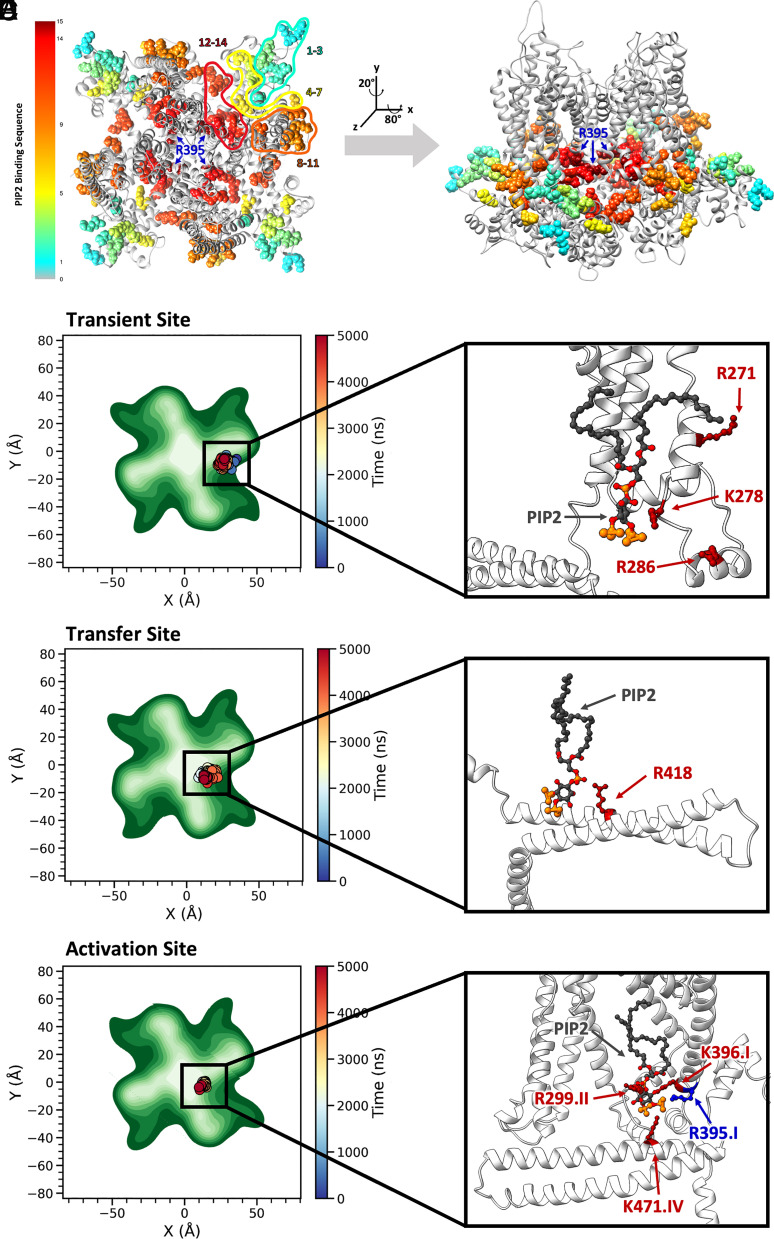
Small-conductance Ca2+-activated K+ channels (SK, KCa2) are gated solely by intracellular microdomain Ca2+. The channel has emerged as a therapeutic target for cardiac arrhythmias. Calmodulin (CaM) interacts with the CaM binding domain (CaMBD) of the SK channels, serving as the obligatory Ca2+ sensor to gate the channels. In heterologous expression systems, phosphatidylinositol 4,5-bisphosphate (PIP2) coordinates with CaM in regulating SK channels. However, the roles and mechanisms of PIP2 in regulating SK channels in cardiomyocytes remain unknown. Here, optogenetics, magnetic nanoparticles, combined with Rosetta structural modeling, and molecular dynamics (MD) simulations revealed the atomistic mechanisms of how PIP2 works in concert with Ca2+-CaM in the SK channel activation. Our computational study affords evidence for the critical role of the amino acid residue R395 in the S6 transmembrane segment, which is localized in propinquity to the intracellular hydrophobic gate. This residue forms a salt bridge with residue E398 in the S6 transmembrane segment from the adjacent subunit. Both R395 and E398 are conserved in all known isoforms of SK channels. Our findings suggest that the binding of PIP2 to R395 residue disrupts the R395:E398 salt bridge, increasing the flexibility of the transmembrane segment S6 and the activation of the channel. Importantly, our findings serve as a platform for testing of structural-based drug designs for therapeutic inhibitors and activators of the SK channel family. The study is timely since inhibitors of SK channels are currently in clinical trials to treat atrial arrhythmias.
Keywords: atrial arrhythmias; calmodulin; optogenetics; phosphatidylinositol 4,5-bisphosphate; small conductance Ca2+-activated K+ channel.
Conflict of interest statement
Competing interests statement:The authors declare no competing interest.
Figures

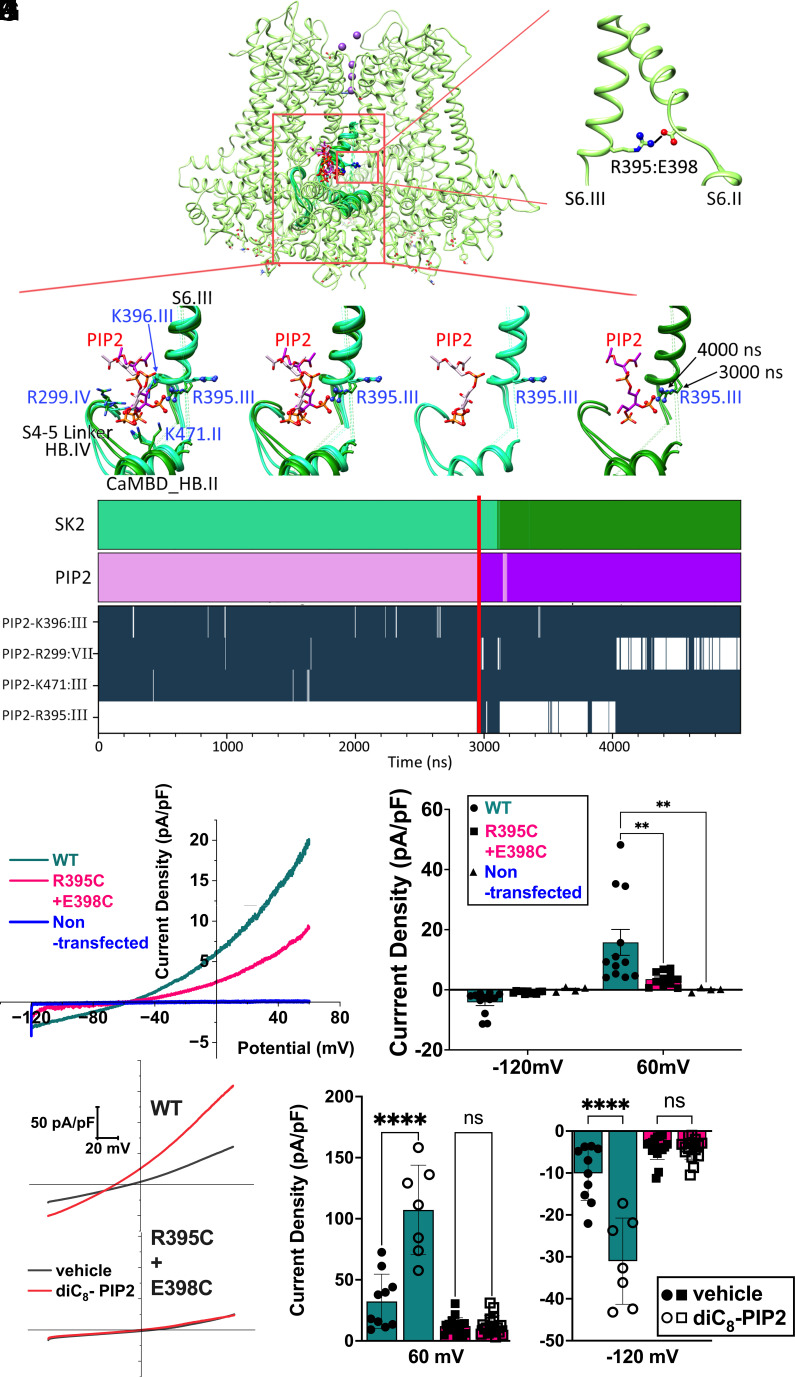
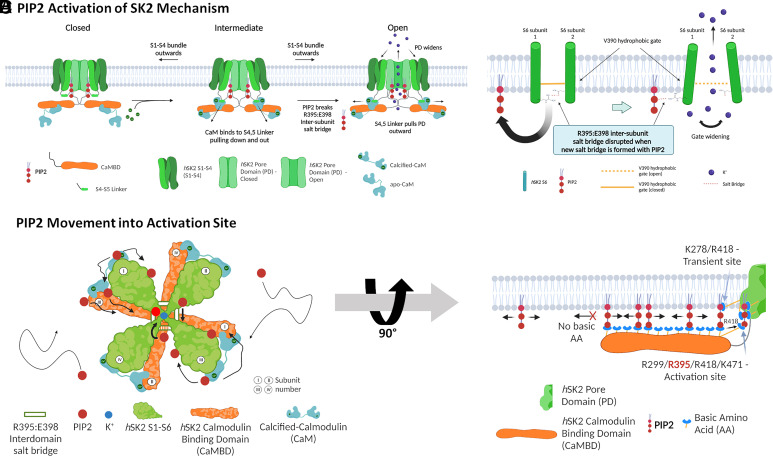
References
-
- Kohler M., et al. , Small-conductance, calcium-activated potassium channels from mammalian brain. Science 273, 1709–1714 (1996). - PubMed
-
- Adelman J. P., Maylie J., Sah P., Small-conductance Ca2+-activated K+ channels: Form and function. Annu. Rev. Physiol. 74, 245–269 (2012). - PubMed
-
- Stocker M., Ca2+-activated K+ channels: Molecular determinants and function of the SK family. Nat. Rev. Neurosci. 5, 758–770 (2004). - PubMed
-
- Strassmaier T., et al. , A novel isoform of SK2 assembles with other SK subunits in mouse brain. J. Biol. Chem. 280, 21231–21236 (2005). - PubMed
MeSH terms
Substances
Grants and funding
- OT2 OD026580/OD/NIH HHS/United States
- NIH R01 DC016099/HHS | NIH | National Institute on Deafness and Other Communication Disorders (NIDCD)
- I01 CX001490/CX/CSRD VA/United States
- T32 GM136597/GM/NIGMS NIH HHS/United States
- R01 DC016099/DC/NIDCD NIH HHS/United States
- NIH F32 HL151130/HHS | NIH | National Heart, Lung, and Blood Institute (NHLBI)
- Anton 2 allocation MCB210014P/Pittsburgh Supercomputing Center
- NIH T32 HL86350/HHS | NIH | National Heart, Lung, and Blood Institute (NHLBI)
- R01 HL158961/HL/NHLBI NIH HHS/United States
- R01 HL137228/HL/NHLBI NIH HHS/United States
- T32 GM007377/GM/NIGMS NIH HHS/United States
- R01 HL174001/HL/NHLBI NIH HHS/United States
- F32 HL151130/HL/NHLBI NIH HHS/United States
- R01 HL128537/HL/NHLBI NIH HHS/United States
- NIH R01 HL085727 NIH R01 HL085844 NIH R01 HL137228/HHS | NIH | National Heart, Lung, and Blood Institute (NHLBI)
- T32 HL086350/HL/NHLBI NIH HHS/United States
- R01 DC004215/DC/NIDCD NIH HHS/United States
- R01 HL152681/HL/NHLBI NIH HHS/United States
- R01 HL085727/HL/NHLBI NIH HHS/United States
- R01 GM116961/GM/NIGMS NIH HHS/United States
- NIH R01 HL152681/HHS | NIH | National Heart, Lung, and Blood Institute (NHLBI)
- R01 AG060504/AG/NIA NIH HHS/United States
- R35 GM149211/GM/NIGMS NIH HHS/United States
- I01 BX000576/BX/BLRD VA/United States
- NIH R01 AG060504 and NIH 2P01 AG051443/HHS | NIH | National Institute on Aging (NIA)
- R01 HL085844/HL/NHLBI NIH HHS/United States
- NIH R01 HL158961/HHS | NIH | National Heart, Lung, and Blood Institute (NHLBI)
- NIH R35 GM149211/HHS | NIH | National Institute of General Medical Sciences (NIGMS)
- P01 AG051443/AG/NIA NIH HHS/United States
- NIH R01 HL128537/HHS | NIH | National Heart, Lung, and Blood Institute (NHLBI)
- R56 HL138392/HL/NHLBI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Miscellaneous
