

Expanding the Spectrum of Endocrine Abnormalities Associated With SOX11-related Disorders
- PMID: 39290158
- PMCID: PMC12168065
- DOI: 10.1210/clinem/dgae620
Expanding the Spectrum of Endocrine Abnormalities Associated With SOX11-related Disorders
Abstract
Context: SOX11 variants cause Coffin-Siris syndrome, characterized by developmental delay, hypogonadotropic hypogonadism, and skeletal and facial defects.
Objective: To examine the contribution of SOX11 variants to the pathogenesis of idiopathic hypogonadotropic hypogonadism (IHH), a disorder caused by hypothalamic GnRH deficiency.
Setting: The Reproductive Endocrine Unit and the Pediatric Endocrinology Division, Massachusetts General Hospital.
Patients or other participants: A cohort of 1810 unrelated IHH probands.
Interventions: Exome sequencing data from the entire cohort were examined for SOX11 rare single nucleotide variants (SNVs) (minor allele frequency in the gnomAD database <0.1%). Rare SOX11 variant association testing was performed between the IHH and gnomAD population. Phenotyping of individuals harboring pathogenic/likely pathogenic SNVs (determined by the American College of Medical Genetics criteria) was performed.
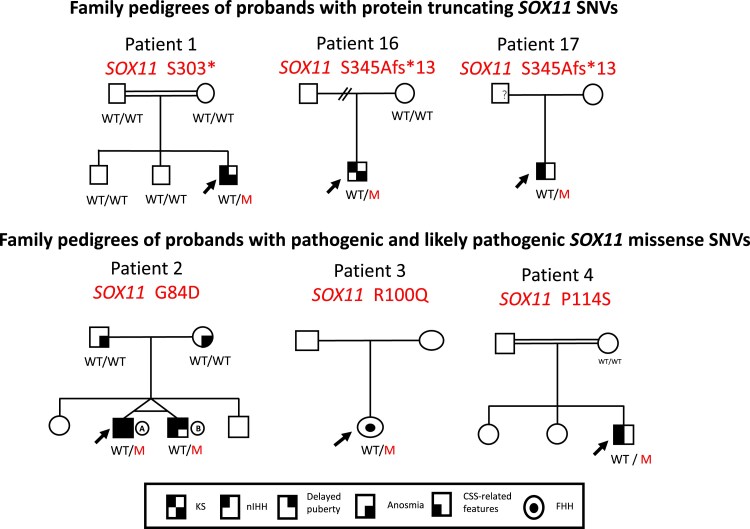
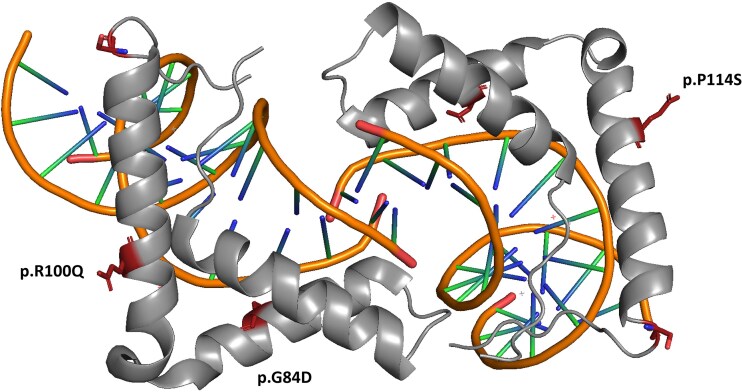
Main outcomes/results: Four pathogenic SOX11 SNVs were identified in 5 IHH probands. The IHH cohort was enriched for SOX11 protein truncating SNVs (frameshift/nonsense) across the entire protein (2 SNVs in 3 IHH cases [p.S303X (de novo); p.S345Afs*13]; P = .0004981) and for SOX11 missense SNVs within the SOX11 high-mobility group domain (2 SNVs in 2 IHH cases p.G84D [de novo]; p.P114S; P = .00313922). The phenotypic spectrum of SOX11 variant carriers revealed additional endocrine defects including anosmic and normosmic forms of IHH, GH deficiency, pituitary and hypothalamic structural defects, and hypothyroidism. A pathogenic SOX11 SNV was also identified in a patient with functional hypogonadotropic hypogonadism (p.R100Q). Coffin-Siris syndrome-associated features were present in 4/5 probands.
Conclusion: Deleterious SOX11 variants cause IHH and other pituitary hormone deficiencies, suggesting that the human SOX11-associated disorder may stem from both hypothalamic and pituitary level defects.
Keywords: SOX11; genetics; hypogonadotropic hypogonadism; hypothalamus; pituitary.
© The Author(s) 2024. Published by Oxford University Press on behalf of the Endocrine Society. All rights reserved. For commercial re-use, please contact reprints@oup.com for reprints and translation rights for reprints. All other permissions can be obtained through our RightsLink service via the Permissions link on the article page on our site—for further information please contact journals.permissions@oup.com. See the journal About page for additional terms.
Figures

References
-
- Reiprich S, Wegner M. From CNS stem cells to neurons and glia: Sox for everyone. Cell Tissue Res. 2015;359(1):111‐124. - PubMed
-
- She ZY, Yang WX. SOX family transcription factors involved in diverse cellular events during development. Eur J Cell Biol. 2015;94(12):547‐563. - PubMed
-
- Berta P, Hawkins JB, Sinclair AH, et al. Genetic evidence equating SRY and the testis-determining factor. Nature. 1990;348(6300):448‐450. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
