

HBV-related HCC development in mice is STAT3 dependent and indicates an oncogenic effect of HBx
- PMID: 39290403
- PMCID: PMC11406364
- DOI: 10.1016/j.jhepr.2024.101128
HBV-related HCC development in mice is STAT3 dependent and indicates an oncogenic effect of HBx
Abstract
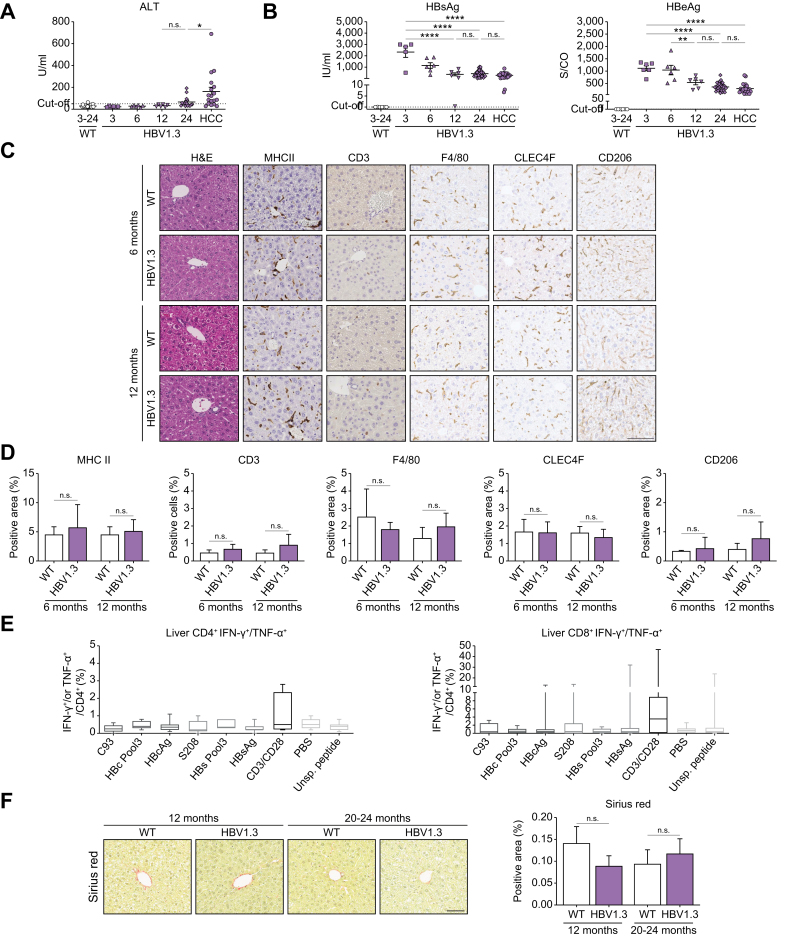
Background & aims: Although most hepatocellular carcinoma (HCC) cases are driven by hepatitis and cirrhosis, a subset of patients with chronic hepatitis B develop HCC in the absence of advanced liver disease, indicating the oncogenic potential of hepatitis B virus (HBV). We investigated the role of HBV transcripts and proteins on HCC development in the absence of inflammation in HBV-transgenic mice.
Methods: HBV-transgenic mice replicating HBV and expressing all HBV proteins from a single integrated 1.3-fold HBV genome in the presence or absence of wild-type HBx (HBV1.3/HBVxfs) were analyzed. Flow cytometry, molecular, histological and in vitro analyses using human cell lines were performed. Hepatocyte-specific Stat3- and Socs3-knockout was analyzed in HBV1.3 mice.
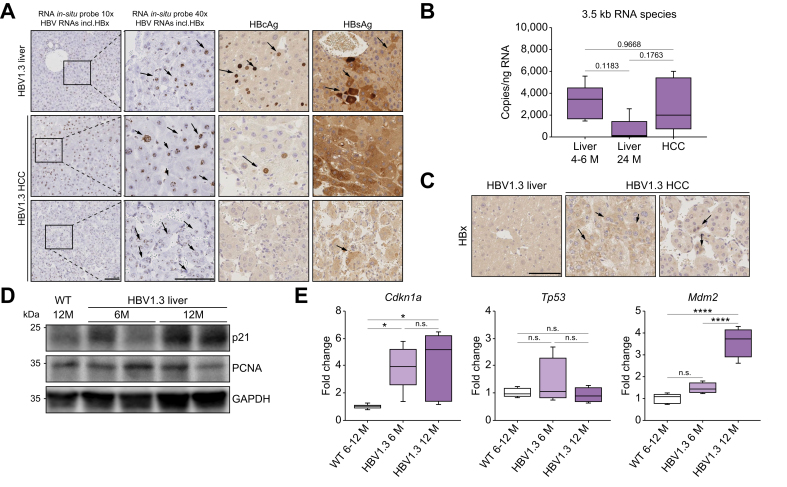
Results: Approximately 38% of HBV1.3 mice developed liver tumors. Protein expression patterns, histology, and mutational landscape analyses indicated that tumors resembled human HCC. HBV1.3 mice showed no signs of active hepatitis, except STAT3 activation, up to the time point of HCC development. HBV-RNAs covering HBx sequence, 3.5-kb HBV RNA and HBx-protein were detected in HCC tissue. Interestingly, HBVxfs mice expressing all HBV proteins except a C-terminally truncated HBx (without the ability to bind DNA damage binding protein 1) showed reduced signs of DNA damage response and had a significantly reduced HCC incidence. Importantly, intercrossing HBV1.3 mice with a hepatocyte-specific STAT3-knockout abrogated HCC development.
Conclusions: Expression of HBV-proteins is sufficient to cause HCC in the absence of detectable inflammation. This indicates the oncogenic potential of HBV and in particular HBx. In our model, HBV-driven HCC was STAT3 dependent. Our study highlights the immediate oncogenic potential of HBV, challenging the idea of a benign highly replicative phase of HBV infection and indicating the necessity for an HBV 'cure'.
Impact and implications: Although most HCC cases in patients with chronic HBV infection occur after a sequence of liver damage and fibrosis, a subset of patients develops HCC without any signs of advanced liver damage. We demonstrate that the expression of all viral transcripts in HBV-transgenic mice suffices to induce HCC development independent of inflammation and fibrosis. These data indicate the direct oncogenic effects of HBV and emphasize the idea of early antiviral treatment in the 'immune-tolerant' phase (HBeAg-positive chronic HBV infection).
Keywords: HBV; HBV x-protein; HBx; HCC; Hepatitis B; Hepatocellular carcinoma; STAT3.
© 2024 The Authors.
Figures





References
-
- Liu Z., Jiang Y., Yuan H., et al. The trends in incidence of primary liver cancer caused by specific etiologies: results from the Global Burden of Disease Study 2016 and implications for liver cancer prevention. J Hepatol. 2019;70:674–683. - PubMed
-
- Trepo C., Chan H.L., Lok A. Hepatitis B virus infection. Lancet. 2014;384:2053–2063. - PubMed
-
- World Health Organization . 2017. Global Hepatitis Report.https://www.who.int/news-room/fact-sheets/detail/hepatitis-b Available from:
-
- Chen C.J., Yang H.I. Natural history of chronic hepatitis B REVEALed. J Gastroenterol Hepatol. 2011;26:628–638. - PubMed
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
