

A comparative analysis of planarian genomes reveals regulatory conservation in the face of rapid structural divergence
- PMID: 39294119
- PMCID: PMC11410931
- DOI: 10.1038/s41467-024-52380-9
A comparative analysis of planarian genomes reveals regulatory conservation in the face of rapid structural divergence
Abstract
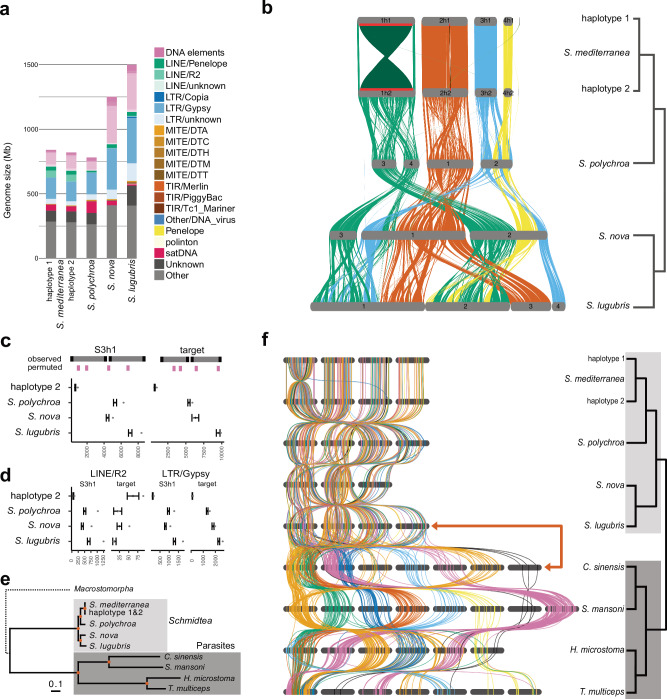
The planarian Schmidtea mediterranea is being studied as a model species for regeneration, but the assembly of planarian genomes remains challenging. Here, we report a high-quality haplotype-phased, chromosome-scale genome assembly of the sexual S2 strain of S. mediterranea and high-quality chromosome-scale assemblies of its three close relatives, S. polychroa, S. nova, and S. lugubris. Using hybrid gene annotations and optimized ATAC-seq and ChIP-seq protocols for regulatory element annotation, we provide valuable genome resources for the planarian research community and a first comparative perspective on planarian genome evolution. Our analyses reveal substantial divergence in protein-coding sequences and regulatory regions but considerable conservation within promoter and enhancer annotations. We also find frequent retrotransposon-associated chromosomal inversions and interchromosomal translocations within the genus Schmidtea and, remarkably, independent and nearly complete losses of ancestral metazoan synteny in Schmidtea and two other flatworm groups. Overall, our results suggest that platyhelminth genomes can evolve without syntenic constraints.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures




References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
