

Genomics 2 Proteins portal: a resource and discovery tool for linking genetic screening outputs to protein sequences and structures
- PMID: 39294369
- PMCID: PMC11466821
- DOI: 10.1038/s41592-024-02409-0
Genomics 2 Proteins portal: a resource and discovery tool for linking genetic screening outputs to protein sequences and structures
Abstract
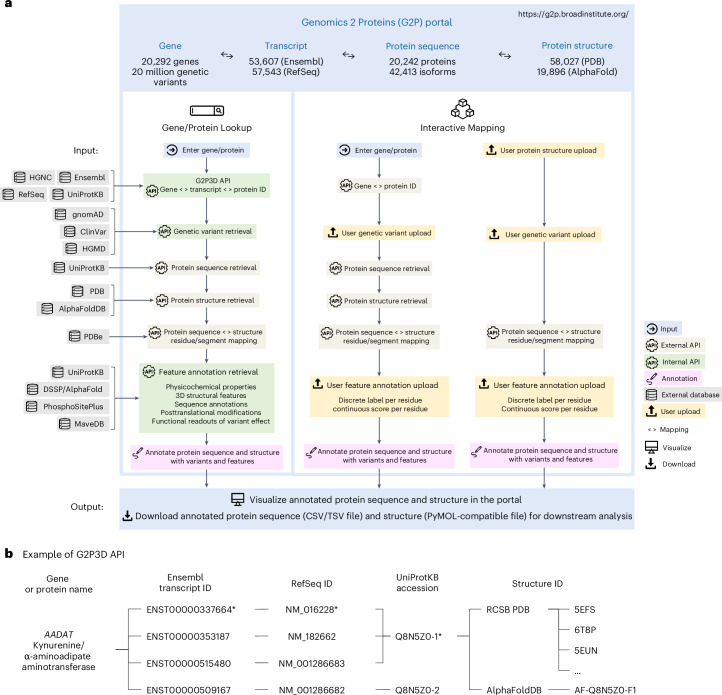
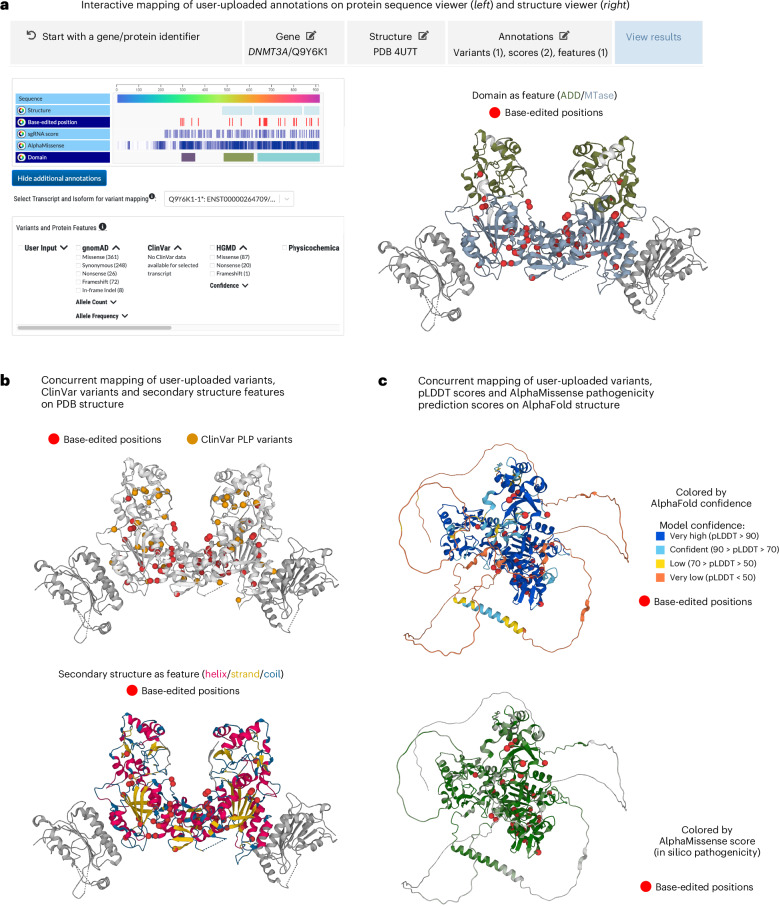
Recent advances in AI-based methods have revolutionized the field of structural biology. Concomitantly, high-throughput sequencing and functional genomics have generated genetic variants at an unprecedented scale. However, efficient tools and resources are needed to link disparate data types-to 'map' variants onto protein structures, to better understand how the variation causes disease, and thereby design therapeutics. Here we present the Genomics 2 Proteins portal ( https://g2p.broadinstitute.org/ ): a human proteome-wide resource that maps 20,076,998 genetic variants onto 42,413 protein sequences and 77,923 structures, with a comprehensive set of structural and functional features. Additionally, the Genomics 2 Proteins portal allows users to interactively upload protein residue-wise annotations (for example, variants and scores) as well as the protein structure beyond databases to establish the connection between genomics to proteins. The portal serves as an easy-to-use discovery tool for researchers and scientists to hypothesize the structure-function relationship between natural or synthetic variations and their molecular phenotypes.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures







Update of
-
Genomics 2 Proteins portal: A resource and discovery tool for linking genetic screening outputs to protein sequences and structures.bioRxiv [Preprint]. 2024 Jan 2:2024.01.02.573913. doi: 10.1101/2024.01.02.573913. bioRxiv. 2024. Update in: Nat Methods. 2024 Oct;21(10):1947-1957. doi: 10.1038/s41592-024-02409-0. PMID: 38260256 Free PMC article. Updated. Preprint.
Similar articles
-
Genomics 2 Proteins portal: A resource and discovery tool for linking genetic screening outputs to protein sequences and structures.bioRxiv [Preprint]. 2024 Jan 2:2024.01.02.573913. doi: 10.1101/2024.01.02.573913. bioRxiv. 2024. Update in: Nat Methods. 2024 Oct;21(10):1947-1957. doi: 10.1038/s41592-024-02409-0. PMID: 38260256 Free PMC article. Updated. Preprint.
-
PhyreRisk: A Dynamic Web Application to Bridge Genomics, Proteomics and 3D Structural Data to Guide Interpretation of Human Genetic Variants.J Mol Biol. 2019 Jun 14;431(13):2460-2466. doi: 10.1016/j.jmb.2019.04.043. Epub 2019 May 7. J Mol Biol. 2019. PMID: 31075275 Free PMC article.
-
G23D: Online tool for mapping and visualization of genomic variants on 3D protein structures.BMC Genomics. 2016 Aug 26;17(1):681. doi: 10.1186/s12864-016-3028-0. BMC Genomics. 2016. PMID: 27565432 Free PMC article.
-
Next Generation Protein Structure Predictions and Genetic Variant Interpretation.J Mol Biol. 2021 Oct 1;433(20):167180. doi: 10.1016/j.jmb.2021.167180. Epub 2021 Aug 4. J Mol Biol. 2021. PMID: 34358547 Review.
-
Structure-based functional inference in structural genomics.J Struct Funct Genomics. 2003;4(2-3):129-35. doi: 10.1023/a:1026200610644. J Struct Funct Genomics. 2003. PMID: 14649297 Review.
Cited by
-
Databases of ligand-binding pockets and protein-ligand interactions.Comput Struct Biotechnol J. 2024 Mar 24;23:1320-1338. doi: 10.1016/j.csbj.2024.03.015. eCollection 2024 Dec. Comput Struct Biotechnol J. 2024. PMID: 38585646 Free PMC article. Review.
-
Identifying Sex Differences in Lung Adenocarcinoma Using Multi-Omics Integrative Protein Signaling Networks.bioRxiv [Preprint]. 2025 Feb 7:2025.02.03.636354. doi: 10.1101/2025.02.03.636354. bioRxiv. 2025. PMID: 39975108 Free PMC article. Preprint.
-
Mapping MAVE data for use in human genomics applications.Genome Biol. 2025 Jun 25;26(1):179. doi: 10.1186/s13059-025-03647-x. Genome Biol. 2025. PMID: 40563119 Free PMC article.
-
CDKN2A and matrix metalloproteinases: key regulators of cellular senescence in squamous cell carcinoma.Am J Transl Res. 2025 Jun 15;17(6):4573-4589. doi: 10.62347/FKDS7259. eCollection 2025. Am J Transl Res. 2025. PMID: 40672601 Free PMC article.
-
Functional evidence for G6PD variant classification from mutational scanning.bioRxiv [Preprint]. 2025 Aug 15:2025.08.11.669723. doi: 10.1101/2025.08.11.669723. bioRxiv. 2025. PMID: 40832240 Free PMC article. Preprint.
References
MeSH terms
Substances
Grants and funding
- RM1 HG010461/HG/NHGRI NIH HHS/United States
- UM1 HG011969/HG/NHGRI NIH HHS/United States
- UM1HG011969/U.S. Department of Health & Human Services | NIH | National Human Genome Research Institute (NHGRI)
- RM1HG010461/U.S. Department of Health & Human Services | NIH | National Human Genome Research Institute (NHGRI)
LinkOut - more resources
Full Text Sources
