

Protein interactions in human pathogens revealed through deep learning
- PMID: 39294458
- PMCID: PMC11445079
- DOI: 10.1038/s41564-024-01791-x
Protein interactions in human pathogens revealed through deep learning
Abstract
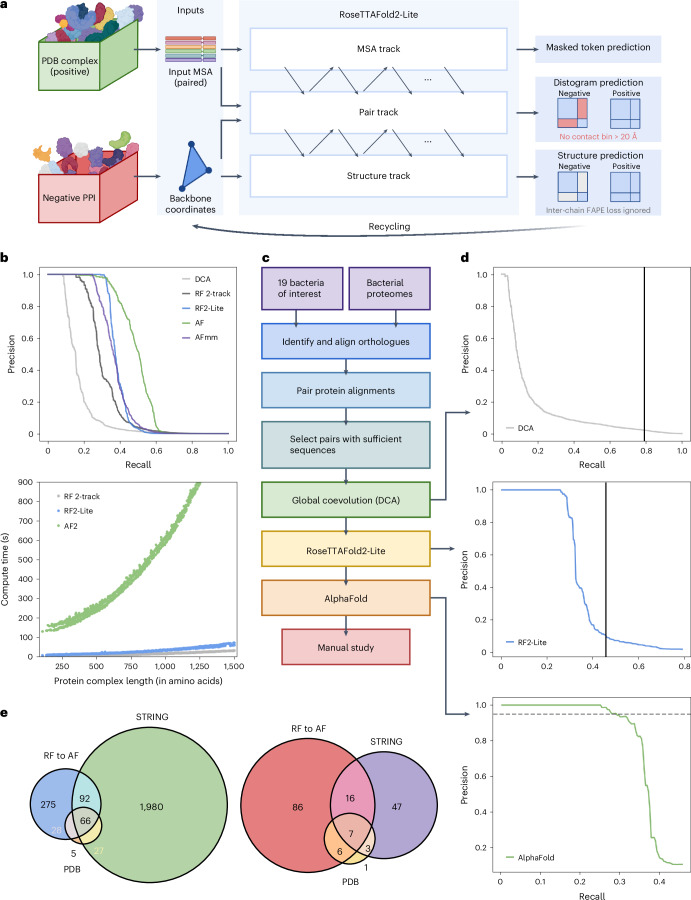
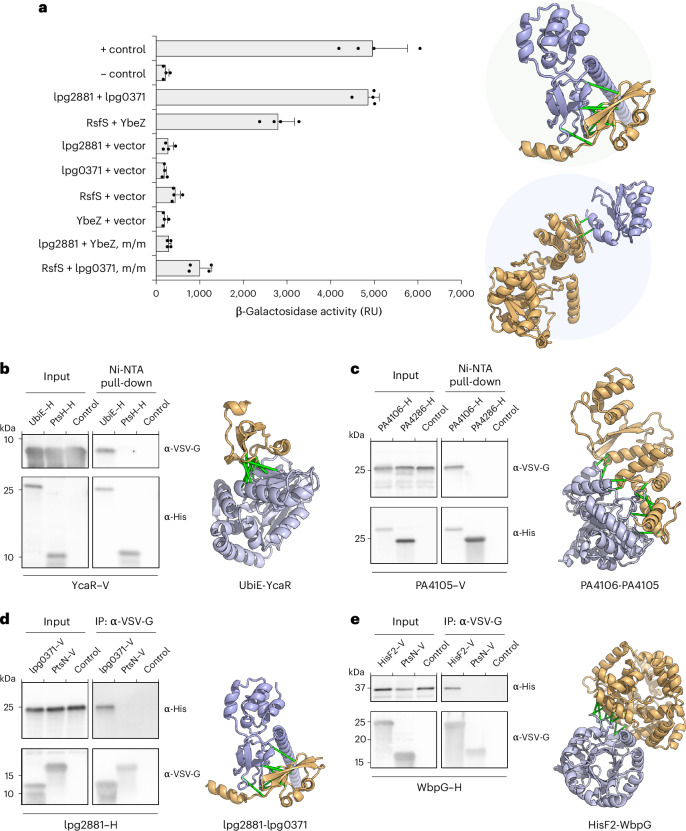
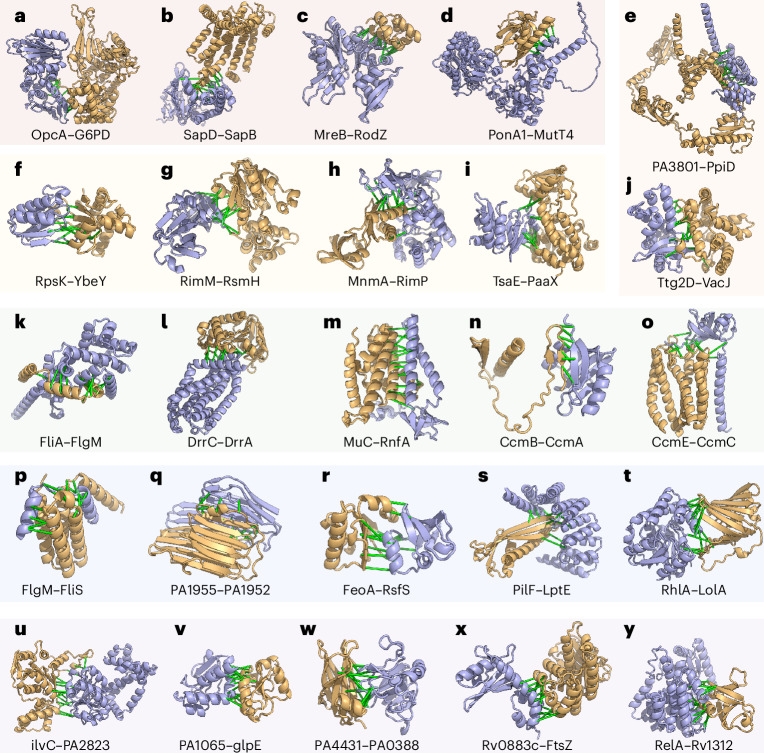
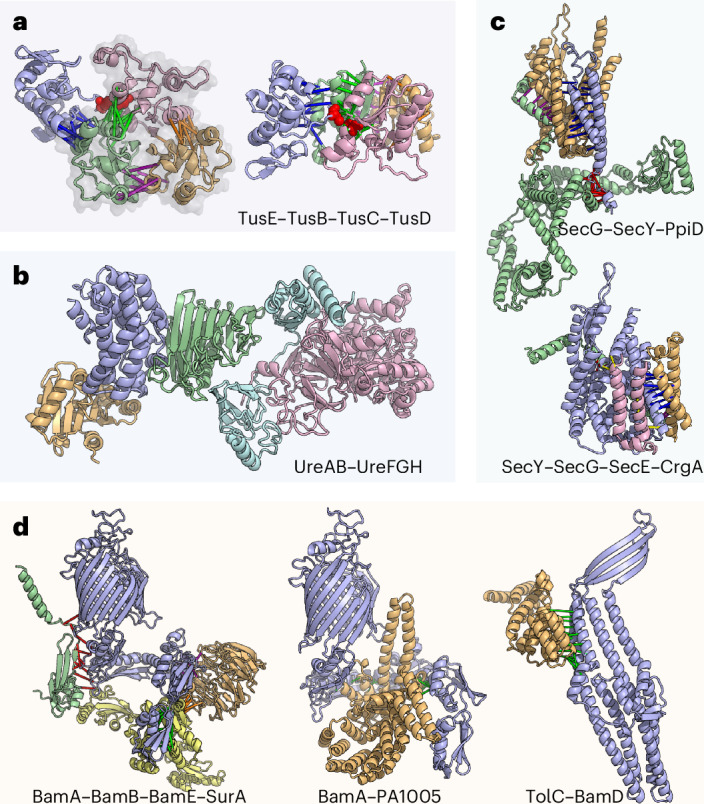
Identification of bacterial protein-protein interactions and predicting the structures of these complexes could aid in the understanding of pathogenicity mechanisms and developing treatments for infectious diseases. Here we developed RoseTTAFold2-Lite, a rapid deep learning model that leverages residue-residue coevolution and protein structure prediction to systematically identify and structurally characterize protein-protein interactions at the proteome-wide scale. Using this pipeline, we searched through 78 million pairs of proteins across 19 human bacterial pathogens and identified 1,923 confidently predicted complexes involving essential genes and 256 involving virulence factors. Many of these complexes were not previously known; we experimentally tested 12 such predictions, and half of them were validated. The predicted interactions span core metabolic and virulence pathways ranging from post-transcriptional modification to acid neutralization to outer-membrane machinery and should contribute to our understanding of the biology of these important pathogens and the design of drugs to combat them.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
Update of
-
Essential and virulence-related protein interactions of pathogens revealed through deep learning.bioRxiv [Preprint]. 2024 Apr 12:2024.04.12.589144. doi: 10.1101/2024.04.12.589144. bioRxiv. 2024. Update in: Nat Microbiol. 2024 Oct;9(10):2642-2652. doi: 10.1038/s41564-024-01791-x. PMID: 38645026 Free PMC article. Updated. Preprint.
