

High-resolution functional mapping of RAD51C by saturation genome editing
- PMID: 39299233
- PMCID: PMC12147941
- DOI: 10.1016/j.cell.2024.08.039
High-resolution functional mapping of RAD51C by saturation genome editing
Abstract
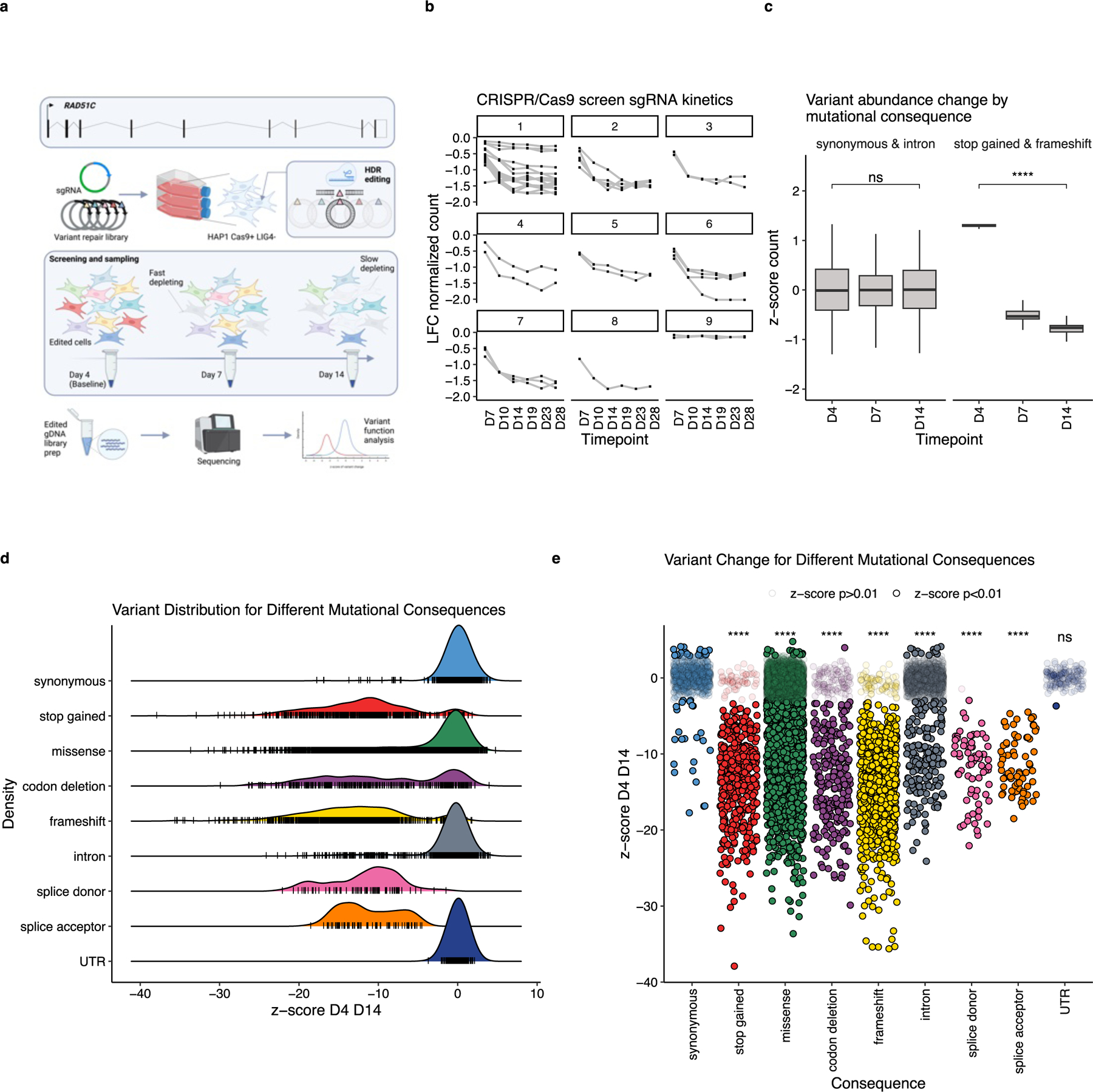
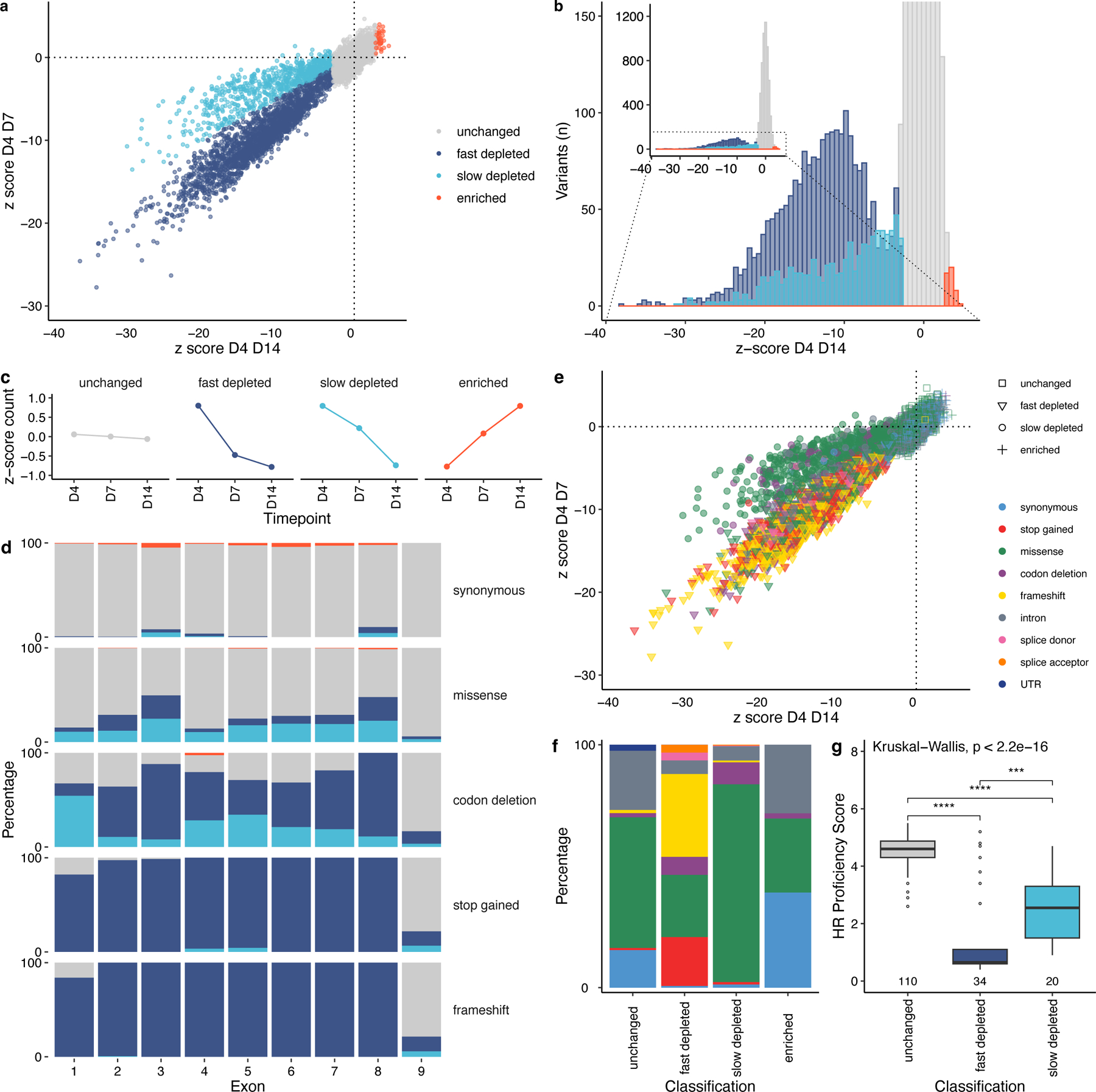
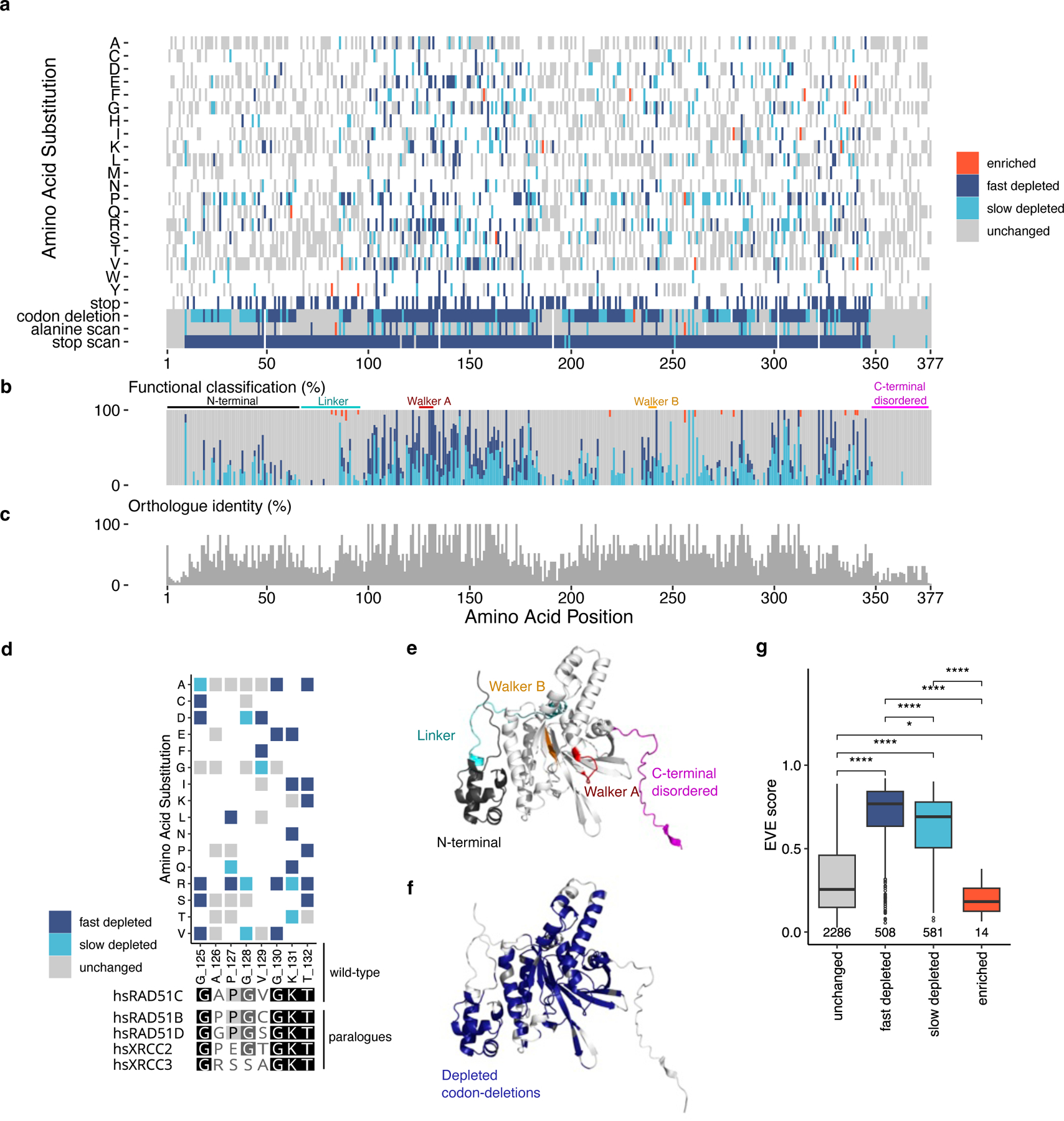
Pathogenic variants in RAD51C confer an elevated risk of breast and ovarian cancer, while individuals homozygous for specific RAD51C alleles may develop Fanconi anemia. Using saturation genome editing (SGE), we functionally assess 9,188 unique variants, including >99.5% of all possible coding sequence single-nucleotide alterations. By computing changes in variant abundance and Gaussian mixture modeling (GMM), we functionally classify 3,094 variants to be disruptive and use clinical truth sets to reveal an accuracy/concordance of variant classification >99.9%. Cell fitness was the primary assay readout allowing us to observe a phenomenon where specific missense variants exhibit distinct depletion kinetics potentially suggesting that they represent hypomorphic alleles. We further explored our exhaustive functional map, revealing critical residues on the RAD51C structure and resolving variants found in cancer-segregating kindred. Furthermore, through interrogation of UK Biobank and a large multi-center ovarian cancer cohort, we find significant associations between SGE-depleted variants and cancer diagnoses.
Keywords: RAD51C; cancer predisposition; homologous recombination; saturation genome editing.
Copyright © 2024 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures


References
-
- Chang TC, Xu K, Cheng Z & Wu G Somatic and Germline Variant Calling from Next-Generation Sequencing Data. in Advances in Experimental Medicine and Biology vol. 1361 37–54 (Springer, 2022). - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
