

Mechanisms of resistance to KRASG12C inhibitors in KRASG12C-mutated non-small cell lung cancer
- PMID: 39301544
- PMCID: PMC11410594
- DOI: 10.3389/fonc.2024.1328728
Mechanisms of resistance to KRASG12C inhibitors in KRASG12C-mutated non-small cell lung cancer
Abstract
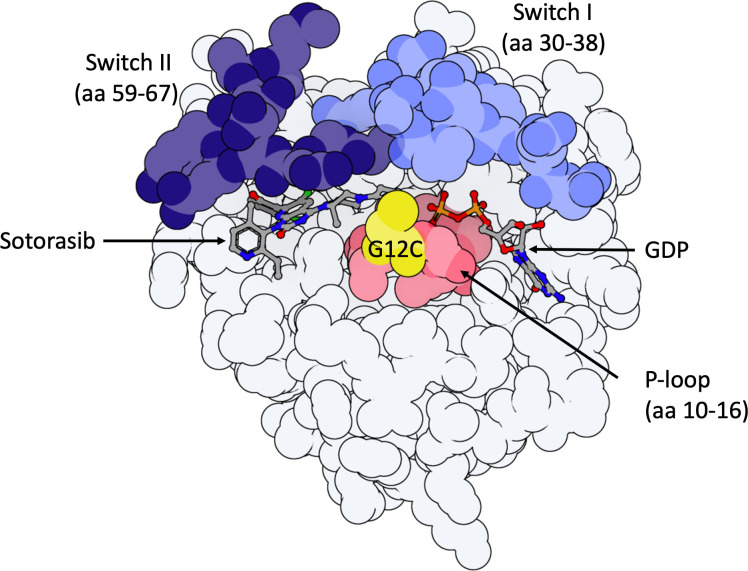
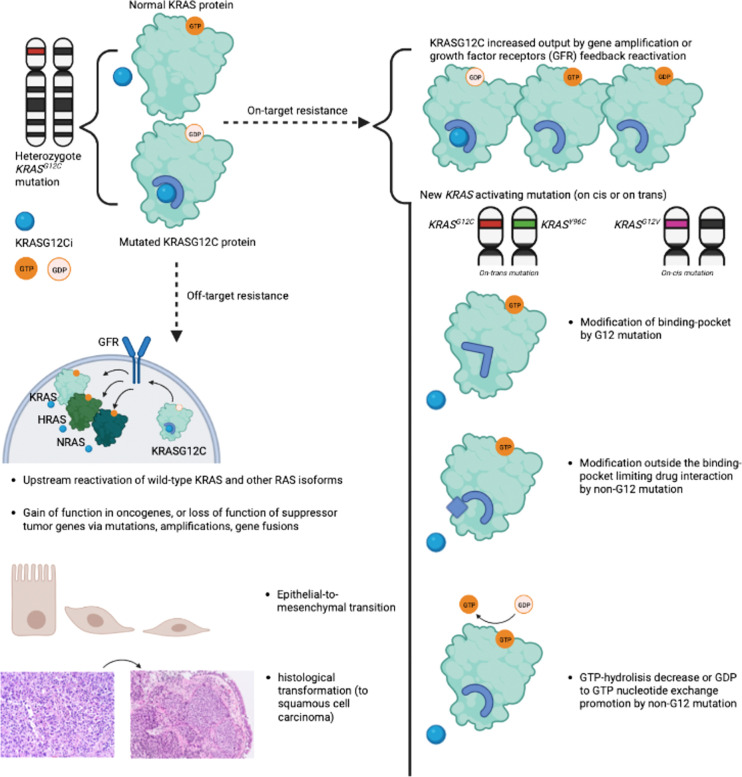
The KRAS protein, a product of the KRAS gene (V-ki-ras2 Kirsten rat sarcoma viral oncogene homolog), functions as a small GTPase that alternates between an active GTP-bound state (KRAS(ON)) and an inactive GDP-bound state (KRAS(OFF)). The KRASG12C mutation results in the accumulation of KRASG12C(OFF), promoting cell cycle survival and proliferation primarily through the canonical MAPK and PI3K pathways. The KRASG12C mutation is found in 13% of lung adenocarcinomas. Previously considered undruggable, sotorasib and adagrasib are the first available OFF-state KRASG12C inhibitors, but treatment resistance is frequent. In this review, after briefly summarizing the KRAS pathway and the mechanism of action of OFF-state KRASG12C inhibitors, we discuss primary and acquired resistance mechanisms. Acquired resistance is the most frequent, with "on-target" mechanisms such as a new KRAS mutation preventing inhibitor binding; and "off-target" mechanisms leading to bypass of KRAS through gain-of-function mutations in other oncogenes such as NRAS, BRAF, and RET; or loss-of-function mutations in tumor suppressor genes such as PTEN. Other "off-target" mechanisms described include epithelial-to-mesenchymal transition and histological transformation. Multiple co-existing mechanisms can be found in patients, but few cases have been published. We highlight the lack of data on non-genomic resistance and the need for comprehensive clinical studies exploring histological, genomic, and non-genomic changes at resistance. This knowledge could help foster new treatment initiatives in this challenging context.
Keywords: KRASG12C inhibitor resistance; KRASG12C mutation; adagrasib; non-small cell lung cancer; sotorasib; translational research.
Copyright © 2024 Chour, Toffart, Berton and Duruisseaux.
Conflict of interest statement
A-CT reports personal fees from AMGEN, during the conduct of the study; personal fees and non-financial support from Novartis, personal fees and non-financial support from Vifor Pharma, personal fees from Boehringer Ingelheim, grants, personal fees and non-financial support from Pfizer, personal fees and non-financial support from MSD, personal fees and non-financial support from Takeda, grants, personal fees and non-financial support from Roche, personal fees and non-financial support from Astra Zeneca, personal fees and non-financial support from BMS, personal fees from Takeda, outside the submitted work. MD reports a membership of an advisory council or committee for BMS, GSK, Sanofi, MSD, AstraZeneca, Abbvie, Takeda, Boehringer Ingelheim, Merus, Amgen, Guardant, Pfizer; consulting fees from Roche, BMS, MSD, AstraZeneca, AbbVie, Takeda, Boehringer Ingelheim, Gamamabs Pharma, Pfizer; research grants from Takeda, NanoString, Lilly, Blueprint. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Figures
References
Publication types
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous