

Absolute Binding Free Energies with OneOPES
- PMID: 39302888
- PMCID: PMC11457222
- DOI: 10.1021/acs.jpclett.4c02352
Absolute Binding Free Energies with OneOPES
Abstract
The calculation of absolute binding free energies (ABFEs) for protein-ligand systems has long been a challenge. Recently, refined force fields and algorithms have improved the quality of the ABFE calculations. However, achieving the level of accuracy required to inform drug discovery efforts remains difficult. Here, we present a transferable enhanced sampling strategy to accurately calculate absolute binding free energies using OneOPES with simple geometric collective variables. We tested the strategy on two protein targets, BRD4 and Hsp90, complexed with a total of 17 chemically diverse ligands, including both molecular fragments and drug-like molecules. Our results show that OneOPES accurately predicts protein-ligand binding affinities with a mean unsigned error within 1 kcal mol-1 of experimentally determined free energies, without the need to tailor the collective variables to each system. Furthermore, our strategy effectively samples different ligand binding modes and consistently matches the experimentally determined structures regardless of the initial protein-ligand configuration. Our results suggest that the proposed OneOPES strategy can be used to inform lead optimization campaigns in drug discovery and to study protein-ligand binding and unbinding mechanisms.
Conflict of interest statement
The authors declare no competing financial interest.
Figures


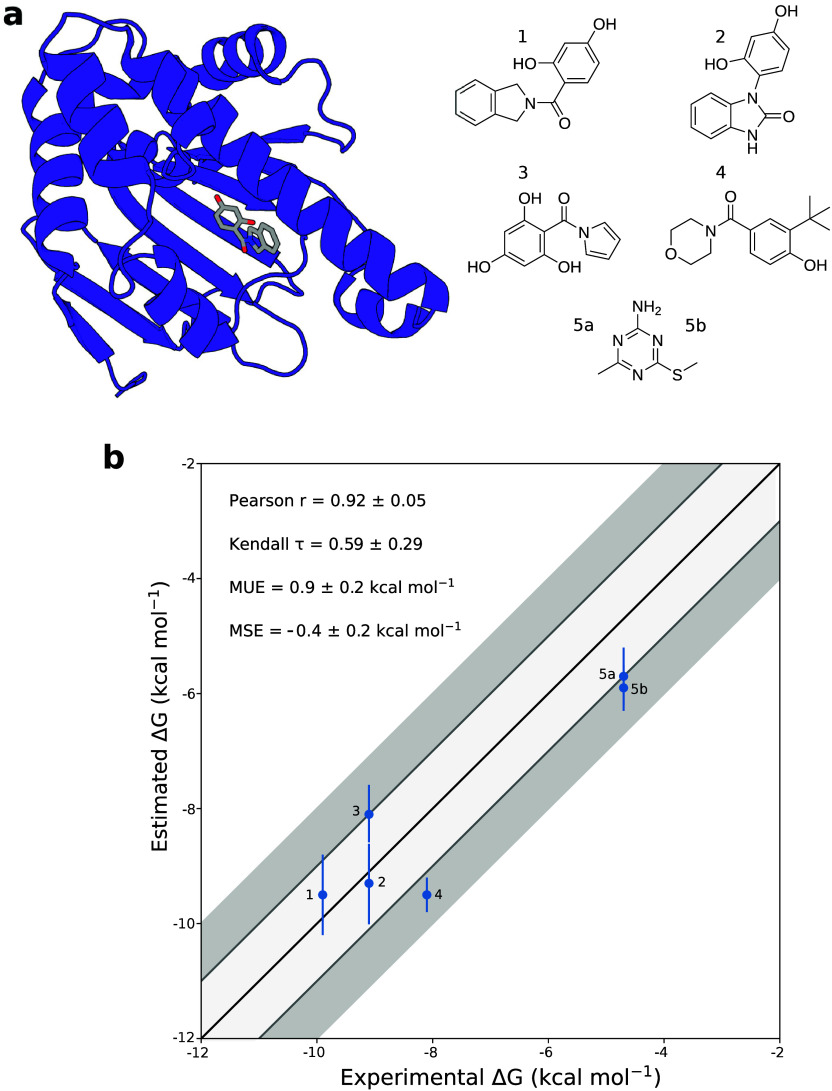
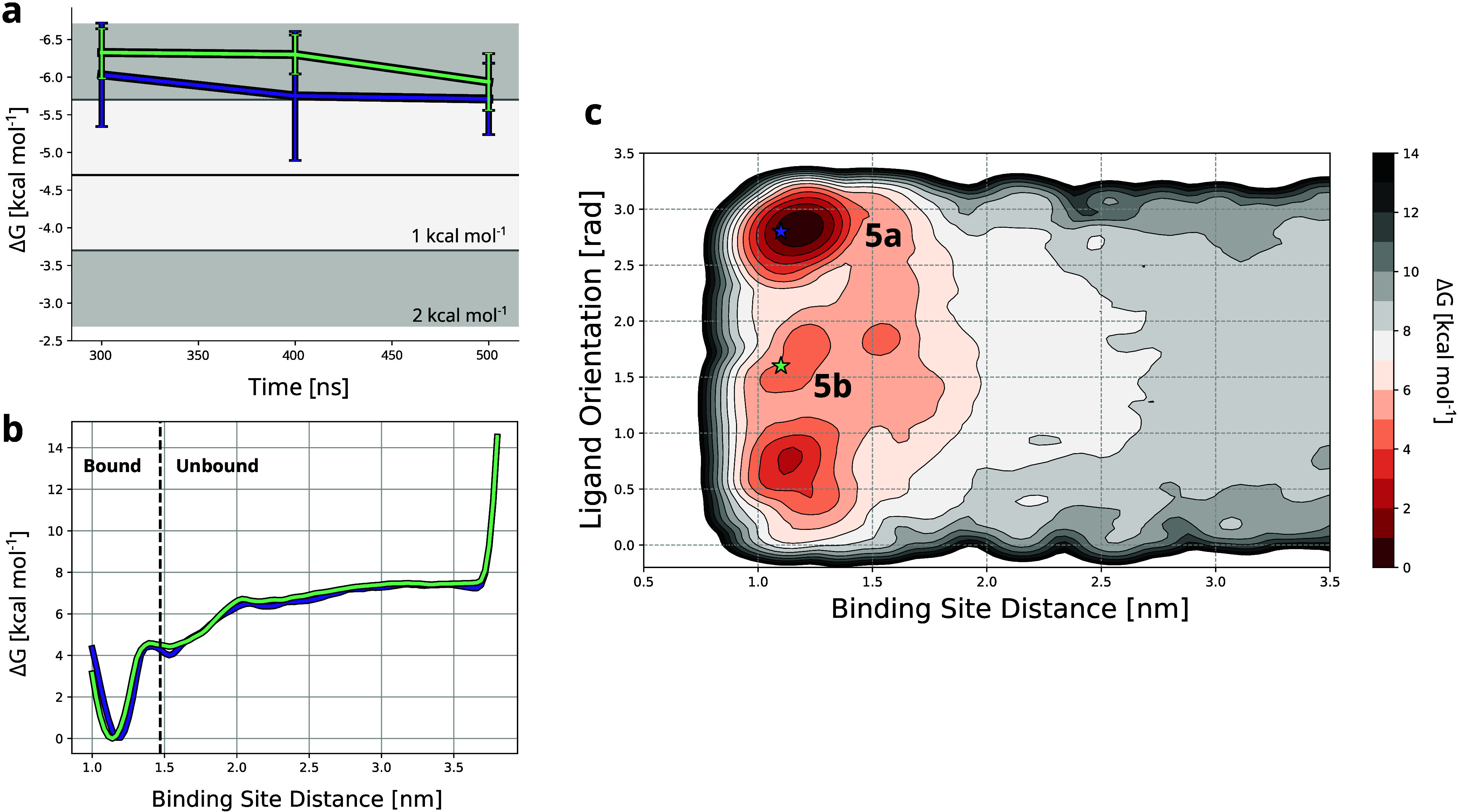
References
-
- Schindler C. E. M.; Baumann H.; Blum A.; Böse D.; Buchstaller H.-P.; Burgdorf L.; Cappel D.; Chekler E.; Czodrowski P.; Dorsch D.; et al. Large-Scale Assessment of Binding Free Energy Calculations in Active Drug Discovery Projects. J. Chem. Inf. Model. 2020, 60, 5457–5474. 10.1021/acs.jcim.0c00900. - DOI - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
