

Defining metabolic flexibility in hair follicle stem cell induced squamous cell carcinoma
- PMID: 39303037
- PMCID: PMC11414736
- DOI: 10.1126/sciadv.adn2806
Defining metabolic flexibility in hair follicle stem cell induced squamous cell carcinoma
Abstract
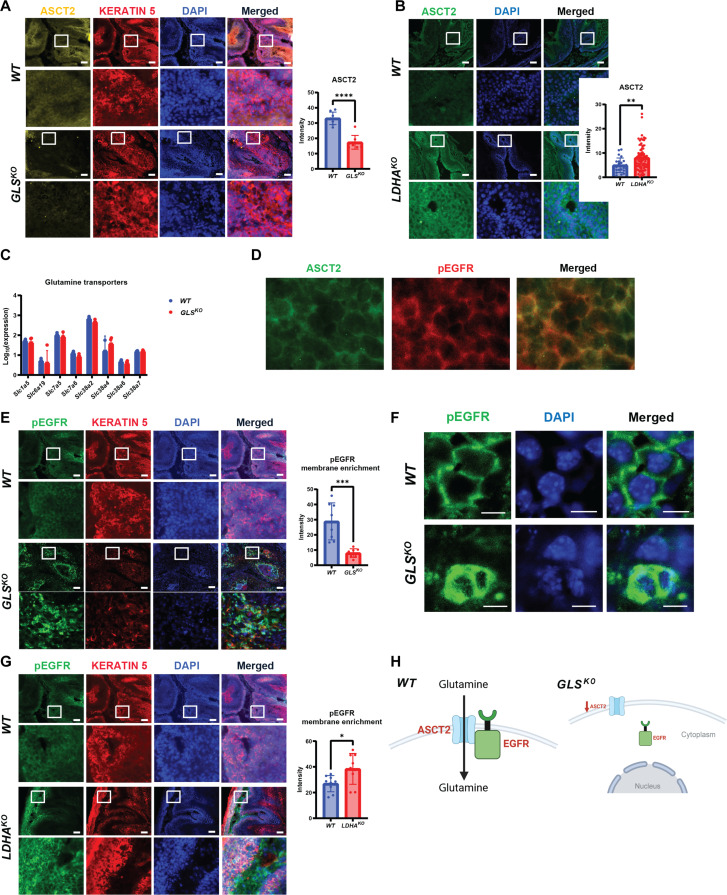
We previously showed that inhibition of glycolysis in cutaneous squamous cell carcinoma (SCC)-initiating cells had no effect on tumorigenesis, despite the perceived requirement of the Warburg effect, which was thought to drive carcinogenesis. Instead, these SCCs were metabolically flexible and sustained growth through glutaminolysis, another metabolic process frequently implicated to fuel tumorigenesis in various cancers. Here, we focused on glutaminolysis and genetically blocked this process through glutaminase (GLS) deletion in SCC cells of origin. Genetic deletion of GLS had little effect on tumorigenesis due to the up-regulated lactate consumption and utilization for the TCA cycle, providing further evidence of metabolic flexibility. We went on to show that posttranscriptional regulation of nutrient transporters appears to mediate metabolic flexibility in this SCC model. To define the limits of this flexibility, we genetically blocked both glycolysis and glutaminolysis simultaneously and found the abrogation of both of these carbon utilization pathways was enough to prevent both papilloma and frank carcinoma.
Figures






Update of
-
Defining metabolic flexibility in hair follicle stem cell induced squamous cell carcinoma.bioRxiv [Preprint]. 2023 Oct 19:2023.10.16.562128. doi: 10.1101/2023.10.16.562128. bioRxiv. 2023. Update in: Sci Adv. 2024 Sep 20;10(38):eadn2806. doi: 10.1126/sciadv.adn2806. PMID: 37905122 Free PMC article. Updated. Preprint.
References
-
- Nassar D., Latil M., Boeckx B., Lambrechts D., Blanpain C., Genomic landscape of carcinogen-induced and genetically induced mouse skin squamous cell carcinoma. Nat. Med. 21, 946–954 (2015). - PubMed
-
- Anderson J. A., Irish J. C., Ngan B. Y., Prevalence of RAS oncogene mutation in head and neck carcinomas. J. Otolaryngol. 21, 321–326 (1992). - PubMed
-
- Pierceall W. E., Mukhopadhyay T., Goldberg L. H., Ananthaswamy H. N., Mutations in the p53 tumor suppressor gene in human cutaneous squamous cell carcinomas. Mol. Carcinog. 4, 445–449 (1991). - PubMed
-
- van der Schroeff J. G., Evers L. M., Boot A. J., Bos J. L., Ras oncogene mutations in basal cell carcinomas and squamous cell carcinomas of human skin. J. Invest. Dermatol. 94, 423–425 (1990). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
