

PRDM16 determines specification of ventricular cardiomyocytes by suppressing alternative cell fates
- PMID: 39304345
- PMCID: PMC11415600
- DOI: 10.26508/lsa.202402719
PRDM16 determines specification of ventricular cardiomyocytes by suppressing alternative cell fates
Abstract
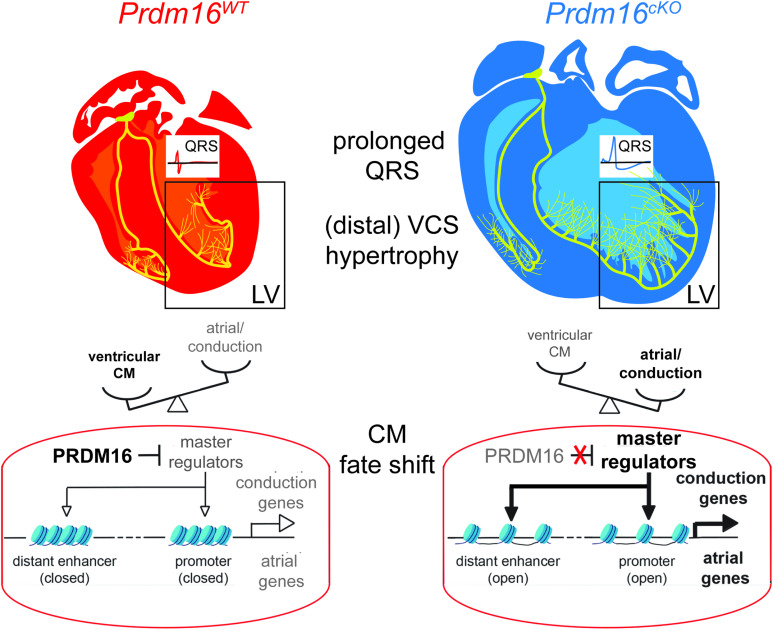
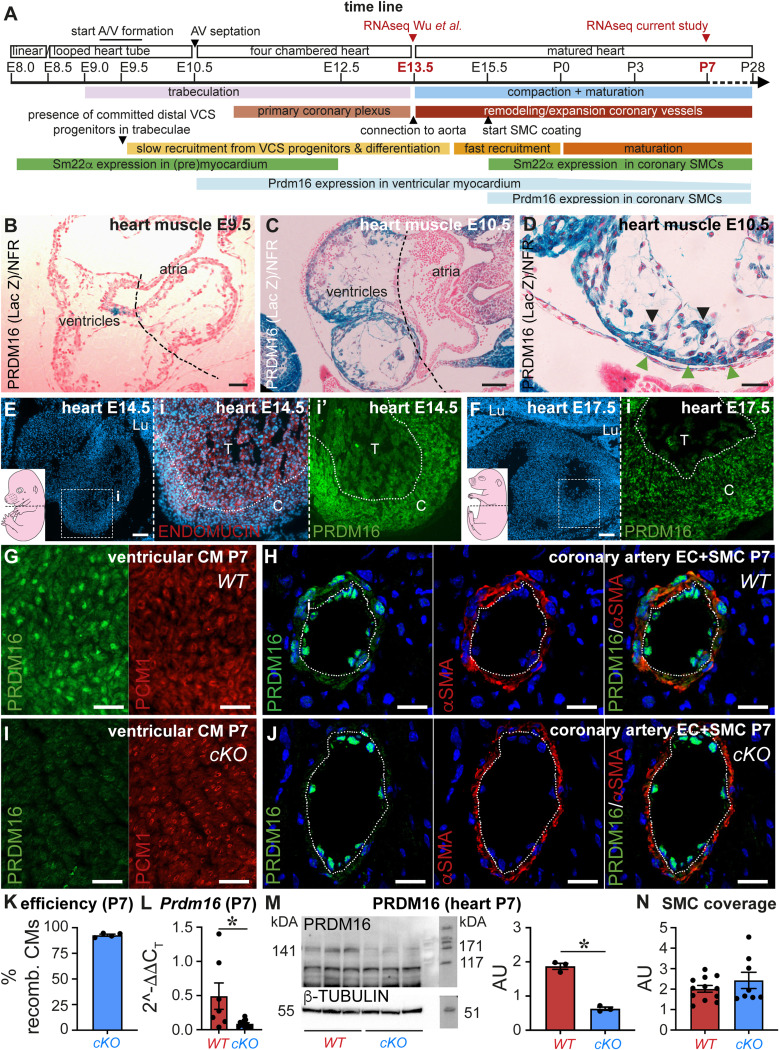
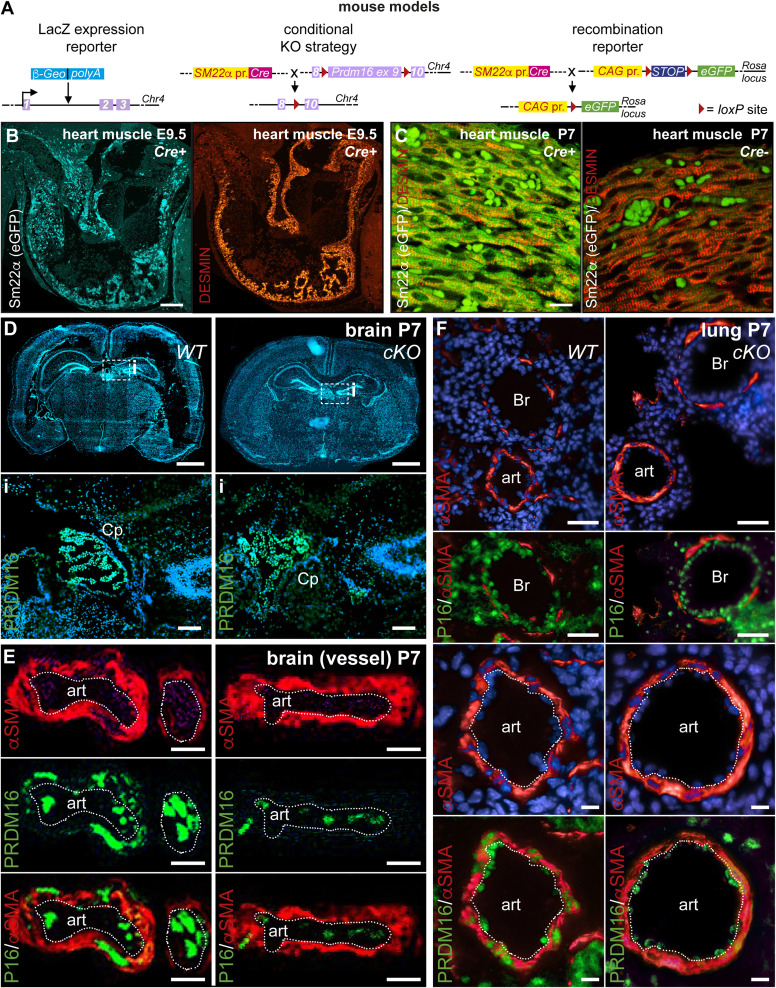
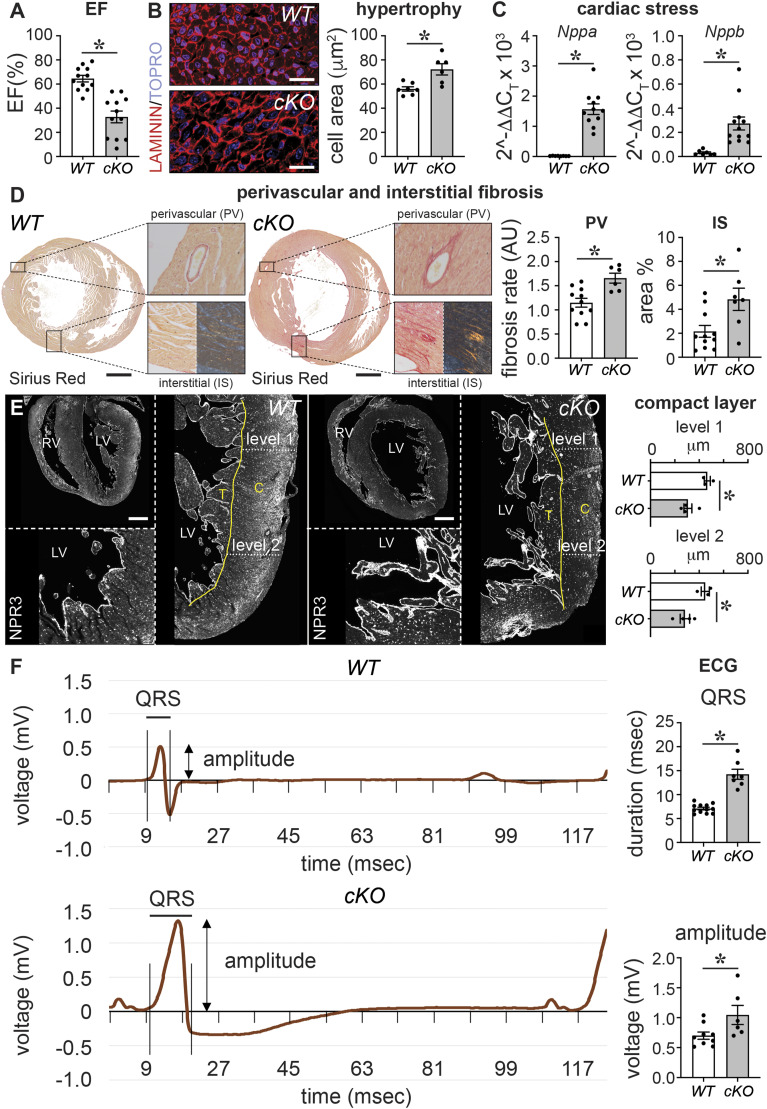
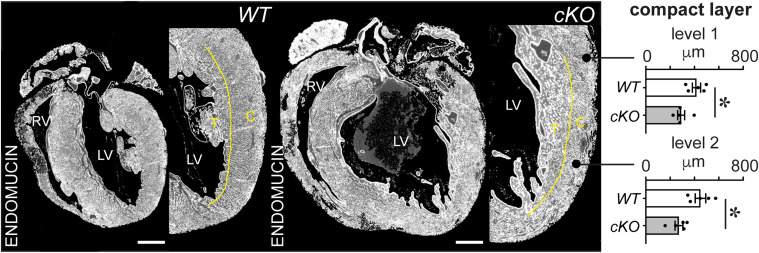
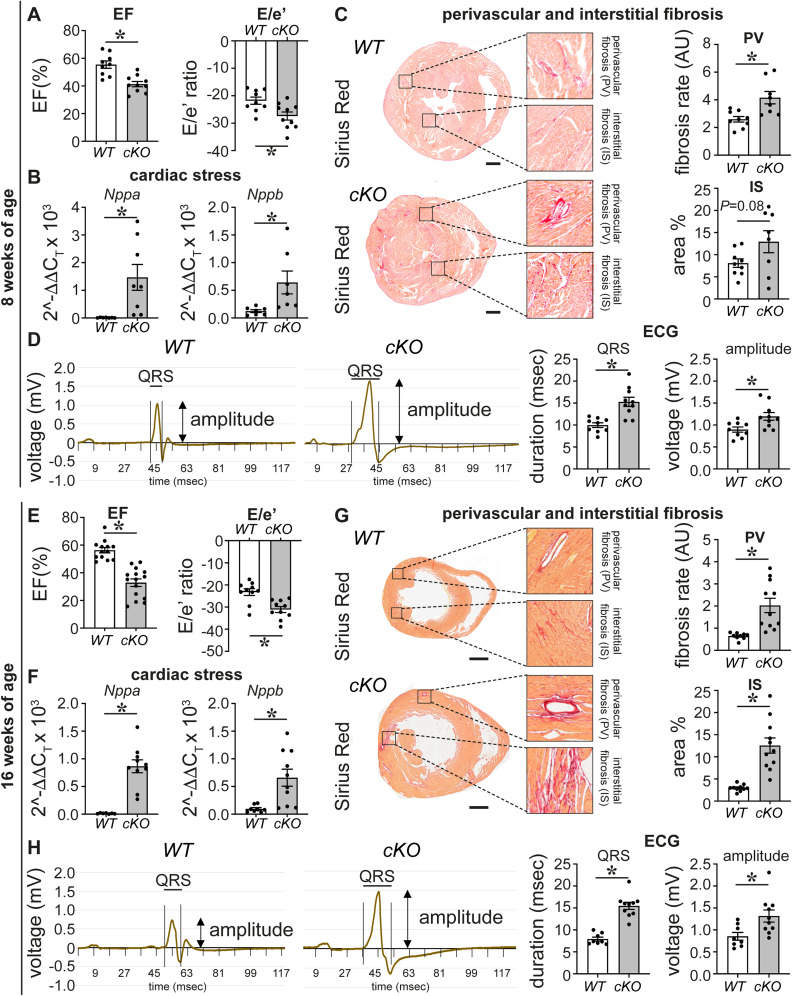
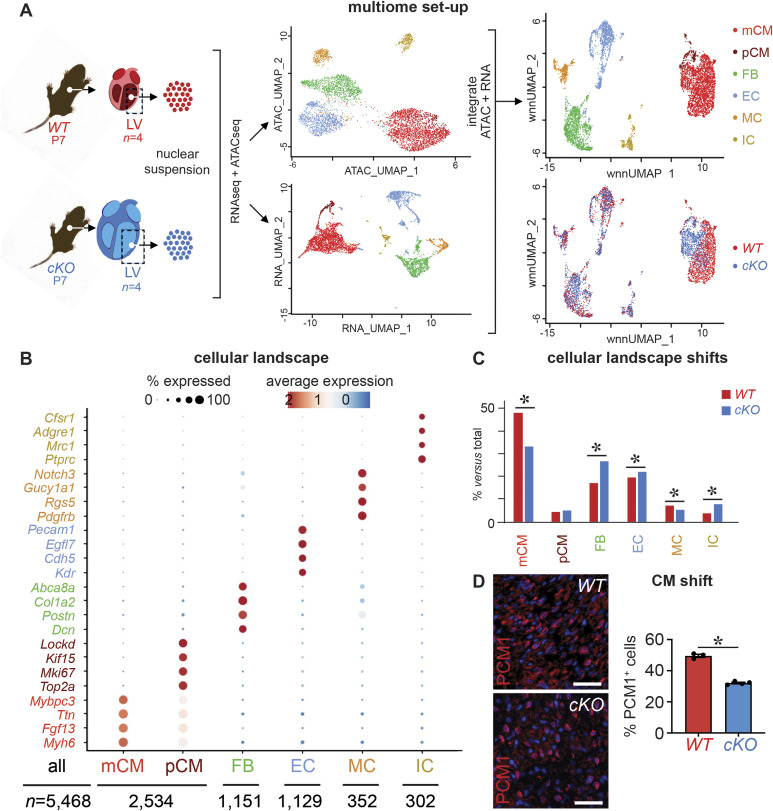
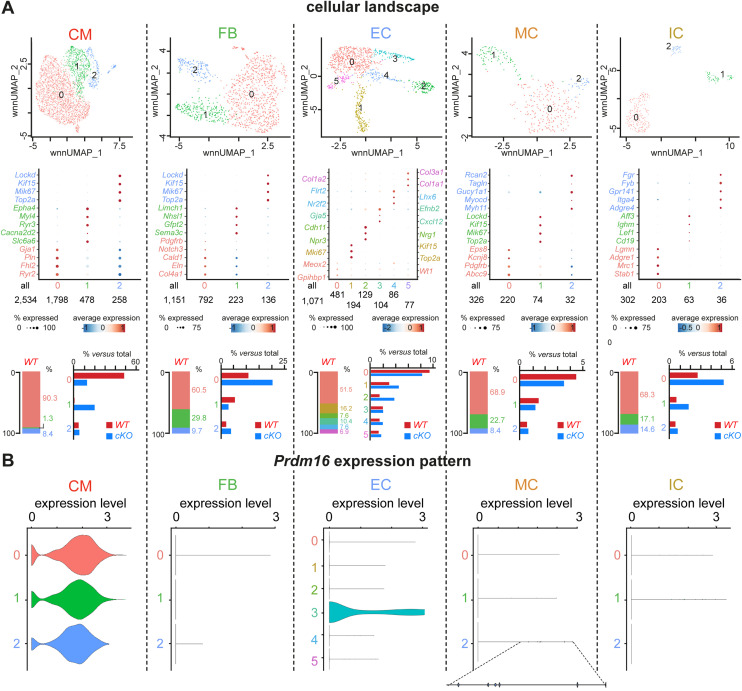
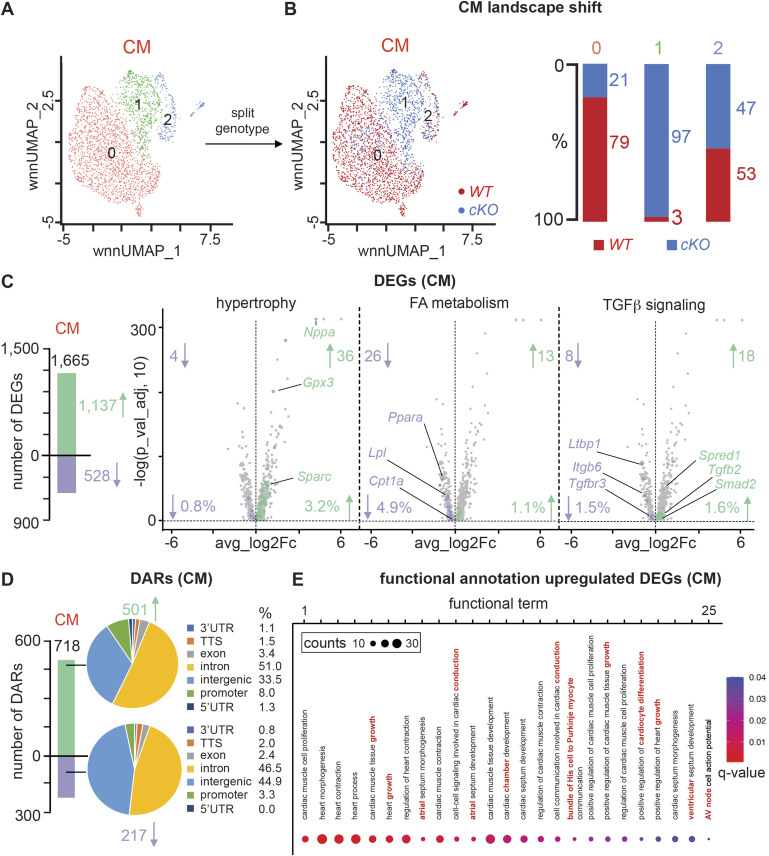
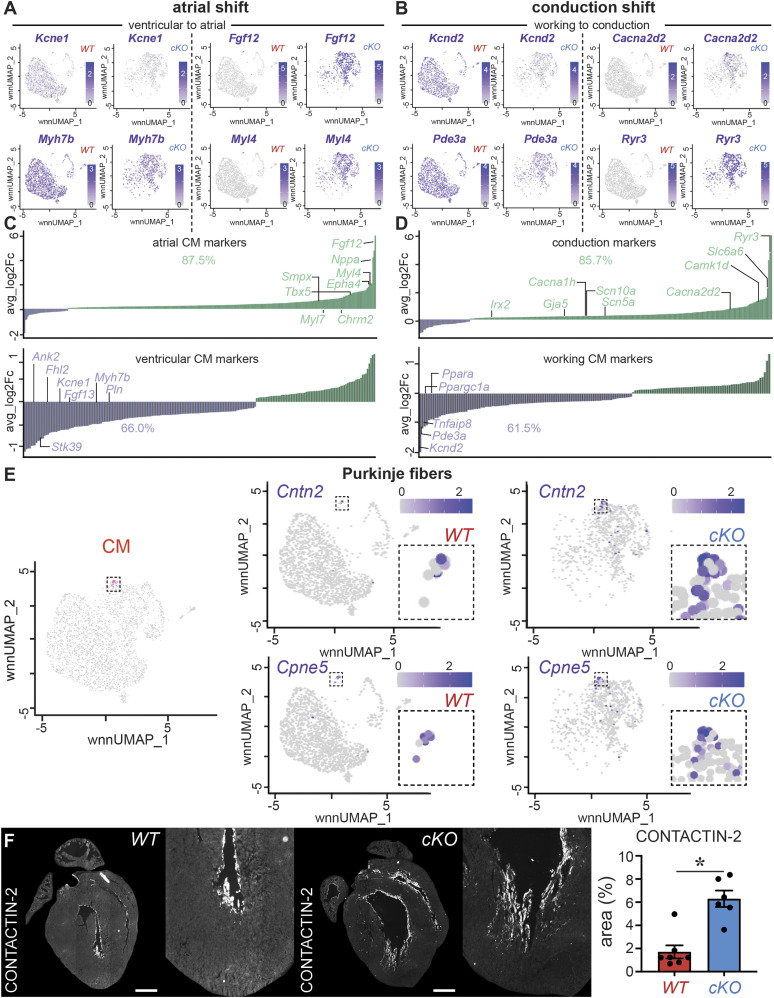
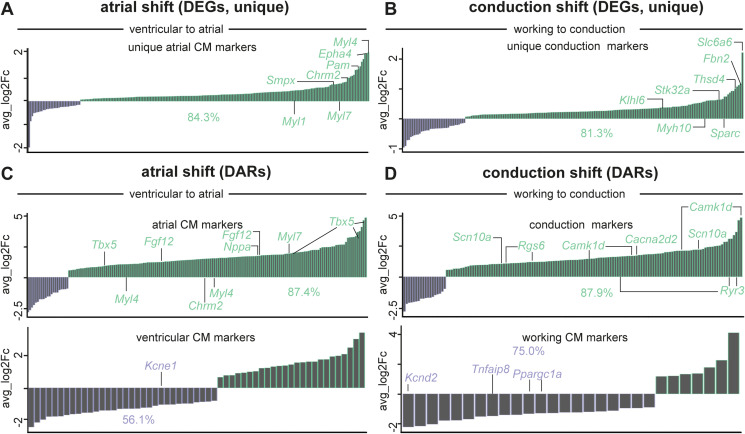
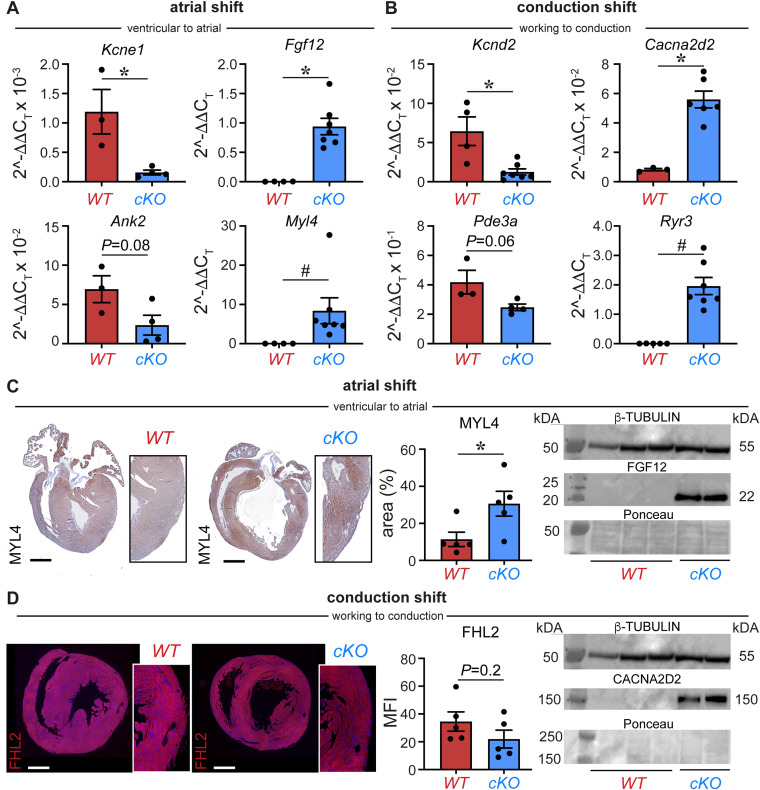
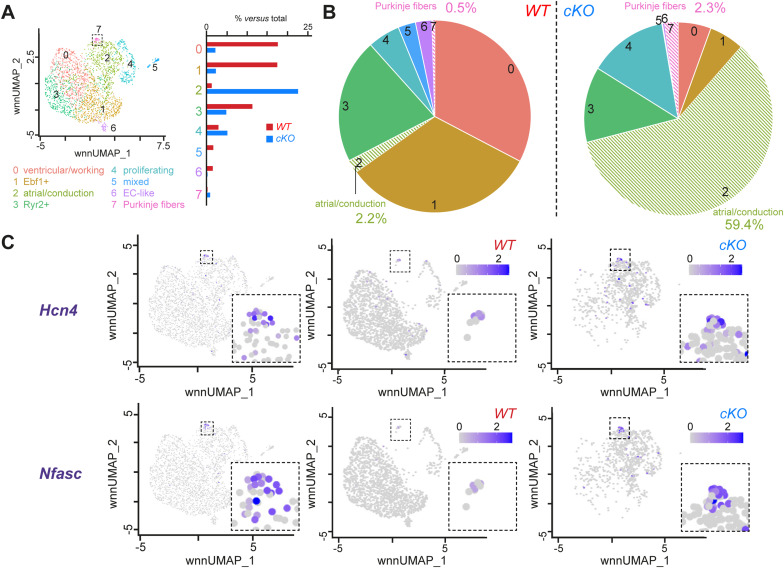
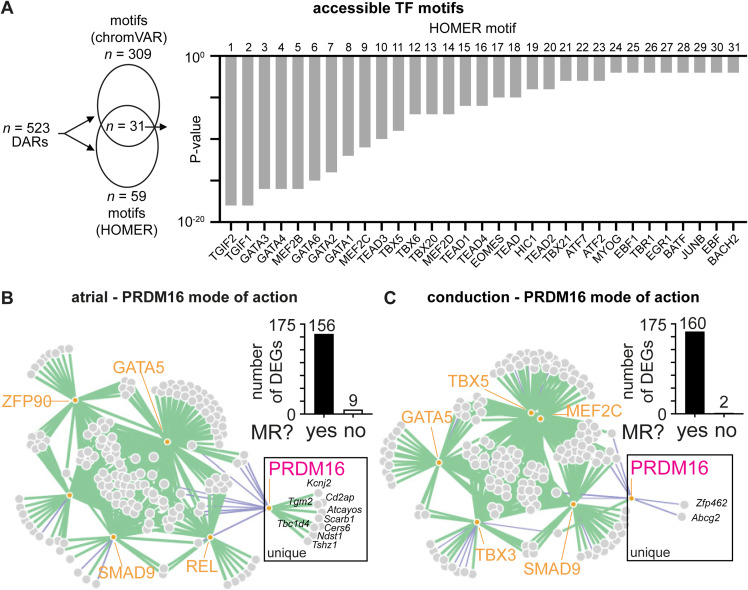
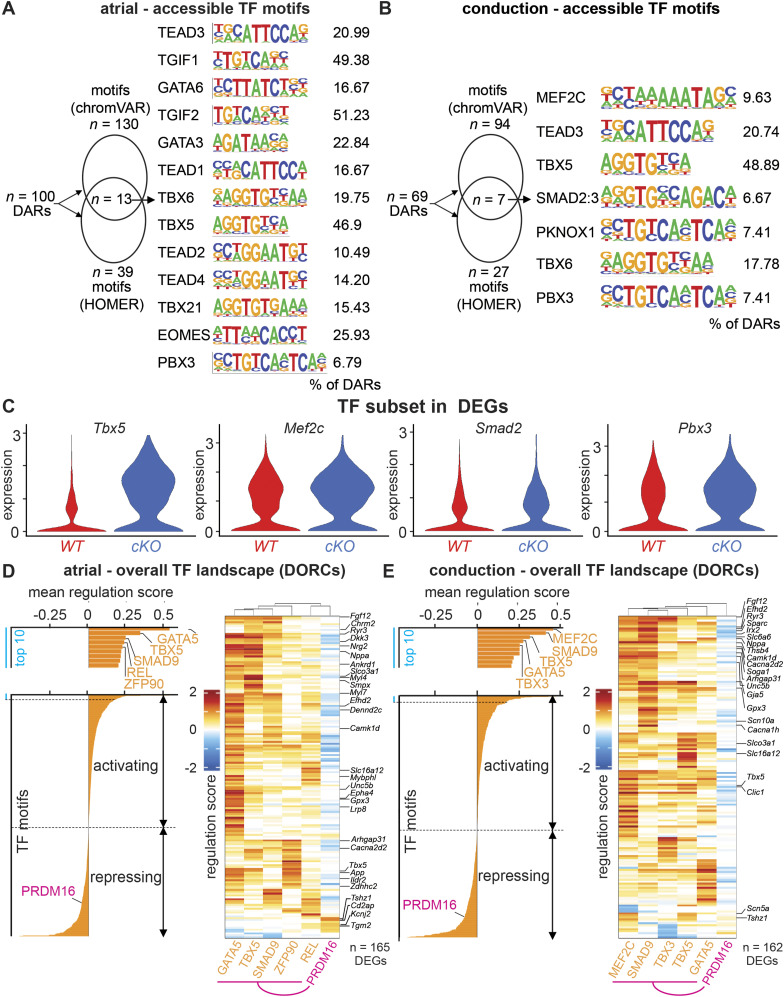
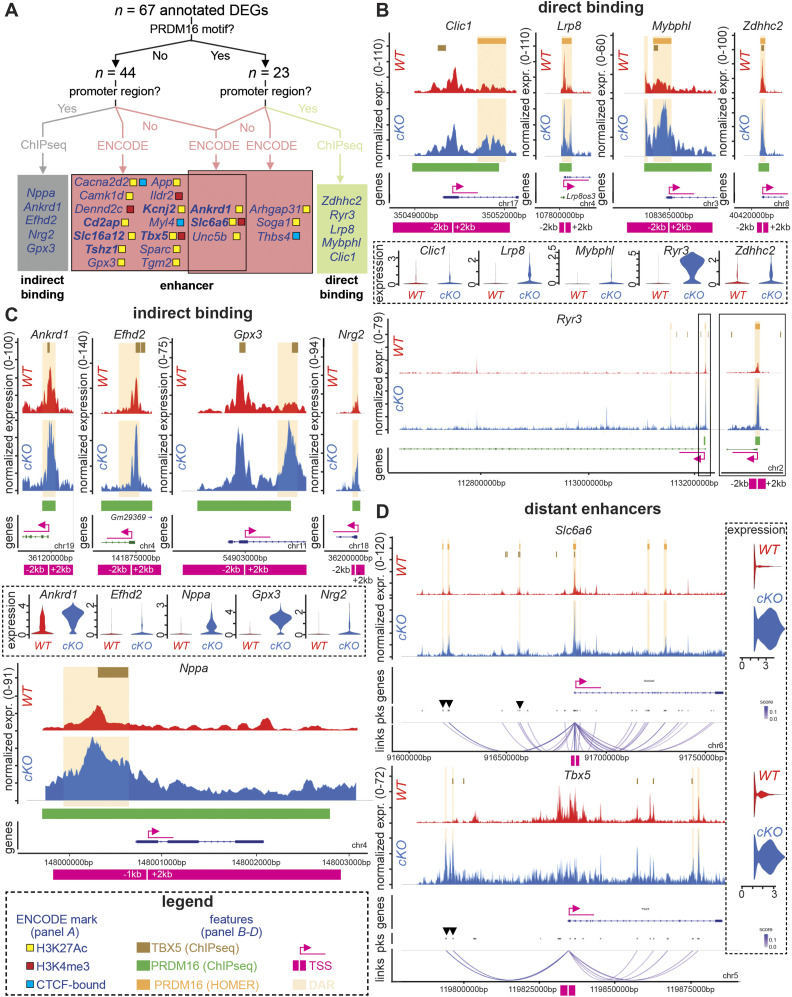
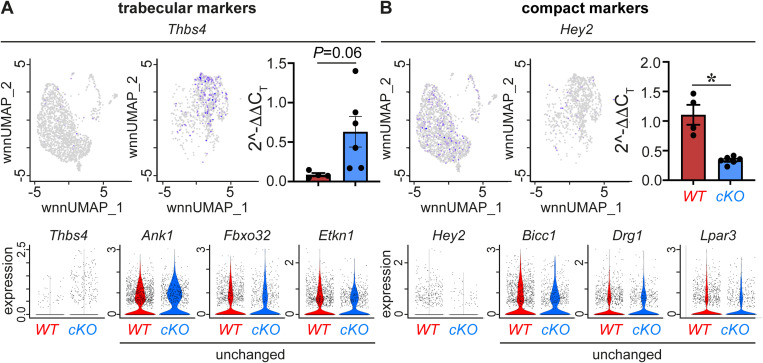
PRDM16 is a transcription factor with histone methyltransferase activity expressed at the earliest stages of cardiac development. Pathogenic mutations in humans lead to cardiomyopathy, conduction abnormalities, and heart failure. PRDM16 is specifically expressed in ventricular but not atrial cardiomyocytes, and its expression declines postnatally. Because in other tissues PRDM16 is best known for its role in binary cell fate decisions, we hypothesized a similar decision-making function in cardiomyocytes. Here, we demonstrated that cardiomyocyte-specific deletion of Prdm16 during cardiac development results in contractile dysfunction and abnormal electrophysiology of the postnatal heart, resulting in premature death. By combined RNA+ATAC single-cell sequencing, we found that PRDM16 favors ventricular working cardiomyocyte identity, by opposing the activity of master regulators of ventricular conduction and atrial fate. Myocardial loss of PRDM16 during development resulted in hyperplasia of the (distal) ventricular conduction system. Hence, PRDM16 plays an indispensable role during cardiac development by driving ventricular working cardiomyocyte identity.
© 2024 Van Wauwe et al.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
References
-
- Amoni M, Vermoortele D, Ekhteraei-Tousi S, Donate Puertas R, Gilbert G, Youness M, Thienpont B, Willems R, Roderick HL, Claus P, et al. (2023) Heterogeneity of repolarization and cell-cell variability of cardiomyocyte remodeling within the myocardial infarction border zone contribute to arrhythmia susceptibility. Circ Arrhythm Electrophysiol 16: e011677. 10.1161/CIRCEP.122.011677 - DOI - PMC - PubMed
-
- Aranguren XL, Agirre X, Beerens M, Coppiello G, Uriz M, Vandersmissen I, Benkheil M, Panadero J, Aguado N, Pascual-Montano A, et al. (2013) Unraveling a novel transcription factor code determining the human arterial-specific endothelial cell signature. Blood 122: 3982–3992. 10.1182/blood-2013-02-483255 - DOI - PubMed
-
- Arndt AK, Schafer S, Drenckhahn JD, Sabeh MK, Plovie ER, Caliebe A, Klopocki E, Musso G, Werdich AA, Kalwa H, et al. (2013) Fine mapping of the 1p36 deletion syndrome identifies mutation of prdm16 as a cause of cardiomyopathy. Am J Hum Genet 93: 67–77. 10.1016/j.ajhg.2013.05.015 - DOI - PMC - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases