

A covalent creatine kinase inhibitor ablates glioblastoma migration and sensitizes tumors to oxidative stress
- PMID: 39304717
- PMCID: PMC11415369
- DOI: 10.1038/s41598-024-73051-1
A covalent creatine kinase inhibitor ablates glioblastoma migration and sensitizes tumors to oxidative stress
Abstract
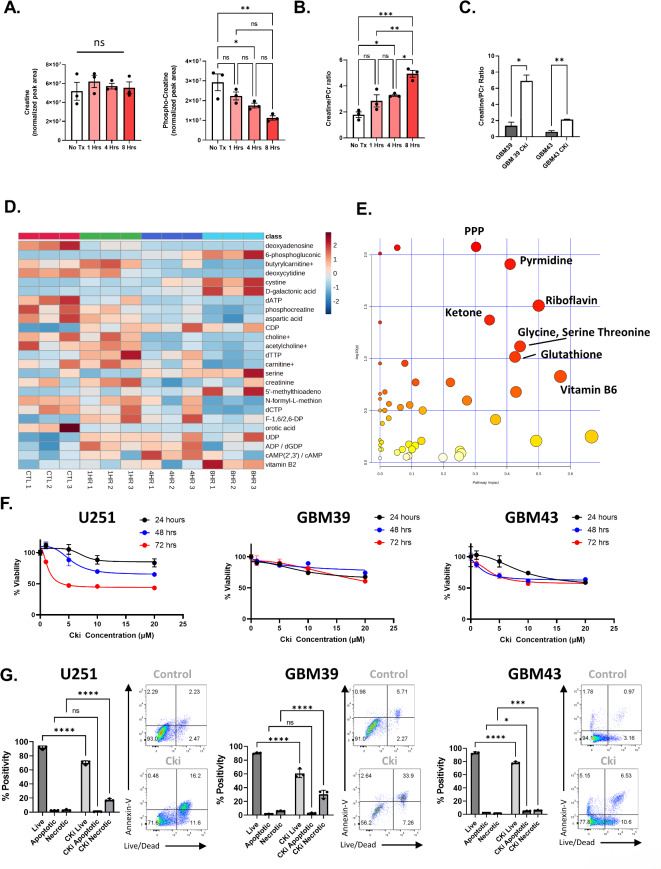
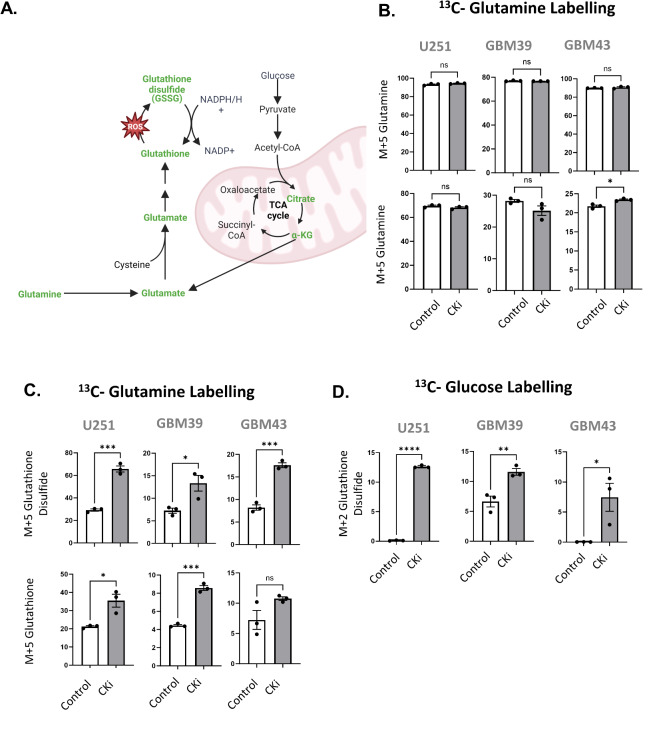
Glioblastoma is a Grade 4 primary brain tumor defined by therapy resistance, diffuse infiltration, and near-uniform lethality. The underlying mechanisms are unknown, and no treatment has been curative. Using a recently developed creatine kinase inhibitor (CKi), we explored the role of this inhibitor on GBM biology in vitro. While CKi minimally impacted GBM cell proliferation and viability, it significantly affected migration. In established GBM cell lines and patient-derived xenografts, CKi ablated both the migration and invasion of GBM cells. CKi also hindered radiation-induced migration. RNA-seq revealed a decrease in invasion-related genes, with an unexpected increase in glutathione metabolism and ferroptosis protection genes post-CKi treatment. The effects of CKi could be reversed by the addition of cell-permeable glutathione. Carbon-13 metabolite tracing indicated heightened glutathione biosynthesis post-CKi treatment. Combinatorial CKi blockade and glutathione inhibition or ferroptosis activation abrogated cell survival. Our data demonstrated that CKi perturbs promigratory and anti-ferroptotic roles in GBM, identifying the creatine kinase axis as a druggable target for GBM treatment.
Keywords: Brain tumor; Cell migration; Enzyme inhibitor; Oxidative stress; Tumor metabolism.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures





References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
