

High-throughput identification of repurposable neuroactive drugs with potent anti-glioblastoma activity
- PMID: 39304781
- PMCID: PMC11564103
- DOI: 10.1038/s41591-024-03224-y
High-throughput identification of repurposable neuroactive drugs with potent anti-glioblastoma activity
Abstract
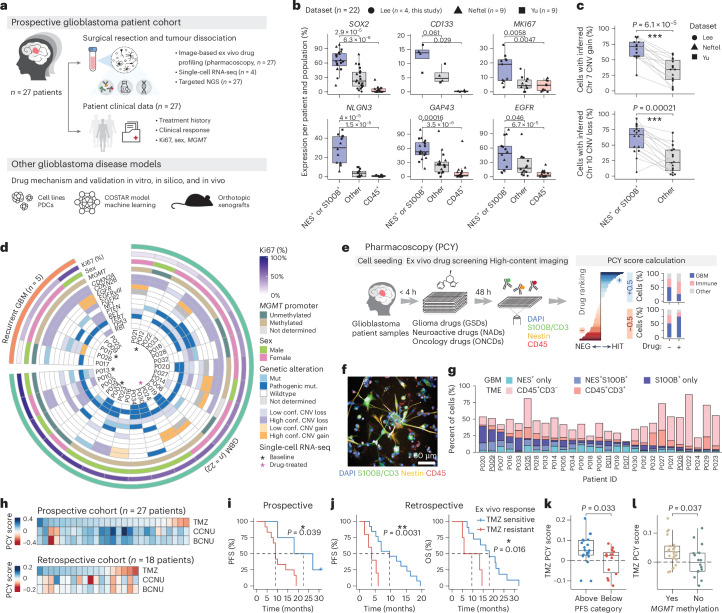
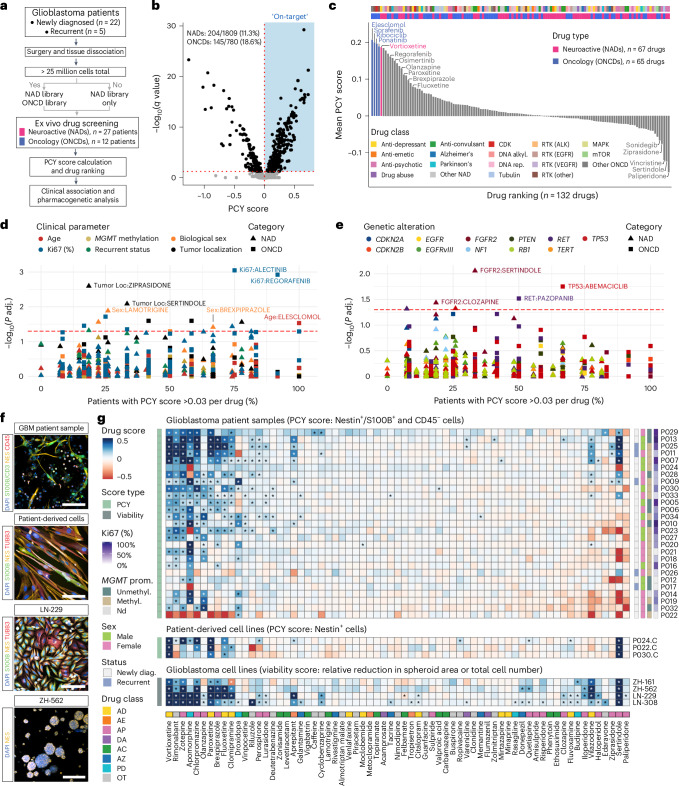
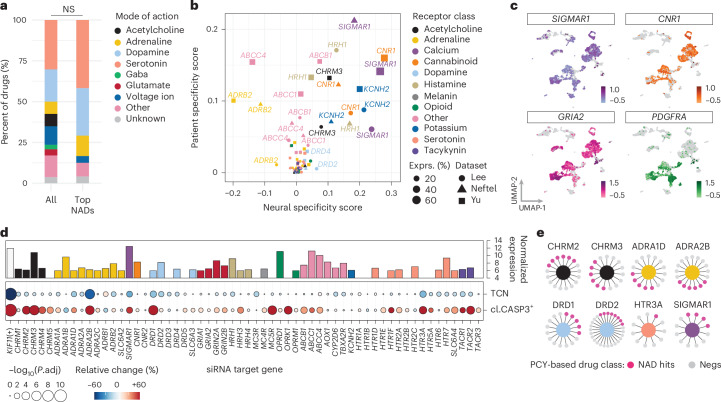
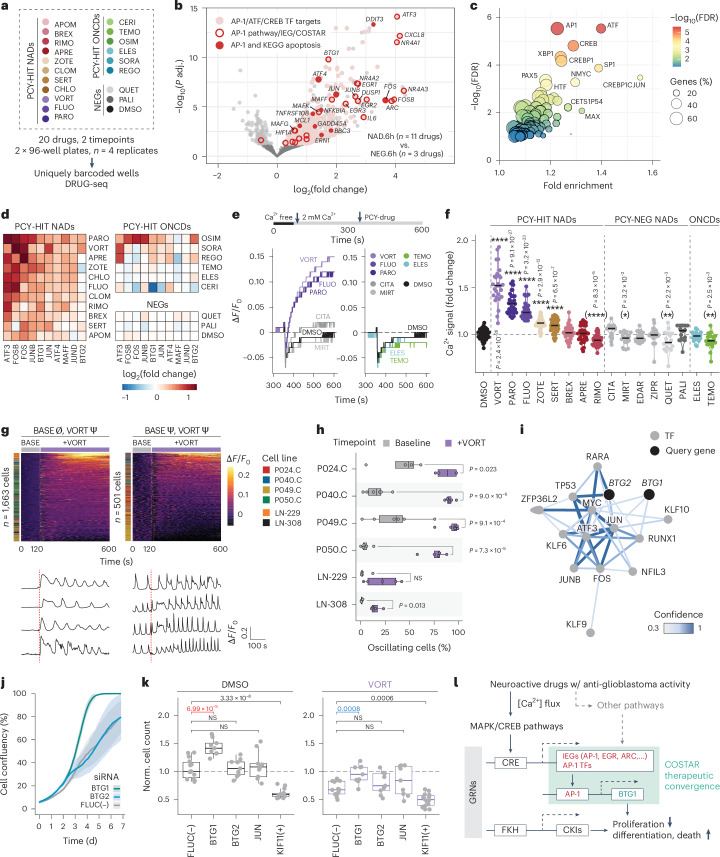
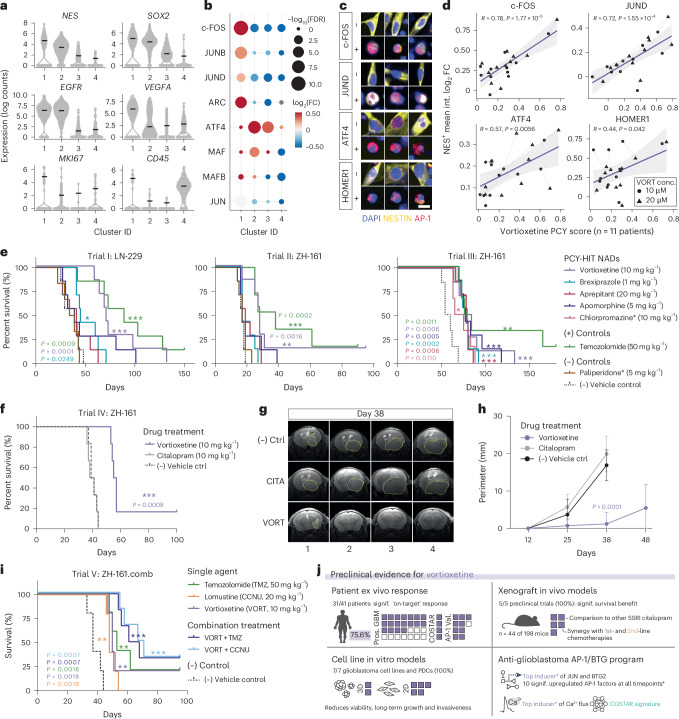
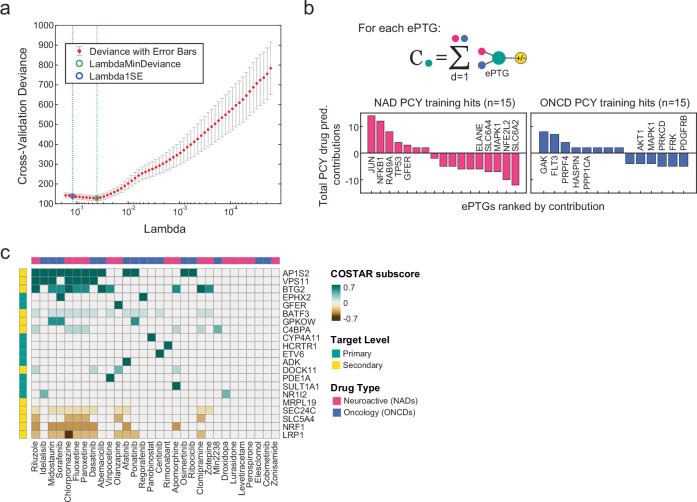
Glioblastoma, the most aggressive primary brain cancer, has a dismal prognosis, yet systemic treatment is limited to DNA-alkylating chemotherapies. New therapeutic strategies may emerge from exploring neurodevelopmental and neurophysiological vulnerabilities of glioblastoma. To this end, we systematically screened repurposable neuroactive drugs in glioblastoma patient surgery material using a clinically concordant and single-cell resolved platform. Profiling more than 2,500 ex vivo drug responses across 27 patients and 132 drugs identified class-diverse neuroactive drugs with potent anti-glioblastoma efficacy that were validated across model systems. Interpretable molecular machine learning of drug-target networks revealed neuroactive convergence on AP-1/BTG-driven glioblastoma suppression, enabling expanded in silico screening of more than 1 million compounds with high patient validation accuracy. Deep multimodal profiling confirmed Ca2+-driven AP-1/BTG-pathway induction as a neuro-oncological glioblastoma vulnerability, epitomized by the anti-depressant vortioxetine synergizing with current standard-of-care chemotherapies in vivo. These findings establish an actionable framework for glioblastoma treatment rooted in its neural etiology.
© 2024. The Author(s).
Conflict of interest statement
Figures










References
MeSH terms
Substances
Grants and funding
- 803063/EC | EU Framework Programme for Research and Innovation H2020 | H2020 Priority Excellent Science | H2020 European Research Council (H2020 Excellent Science - European Research Council)
- 898549/EC | EU Framework Programme for Research and Innovation H2020 | H2020 Priority Excellent Science | H2020 European Research Council (H2020 Excellent Science - European Research Council)
- 956-2019/European Molecular Biology Organization (EMBO)
- 310030_204972/Schweizerischer Nationalfonds zur Förderung der Wissenschaftlichen Forschung (Swiss National Science Foundation)
- 310030_185155/Schweizerischer Nationalfonds zur Förderung der Wissenschaftlichen Forschung (Swiss National Science Foundation)
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
