

AXL and SHC1 confer crizotinib resistance in patient-derived xenograft model of ALK-driven lung cancer
- PMID: 39310759
- PMCID: PMC11416680
- DOI: 10.1016/j.isci.2024.110846
AXL and SHC1 confer crizotinib resistance in patient-derived xenograft model of ALK-driven lung cancer
Abstract
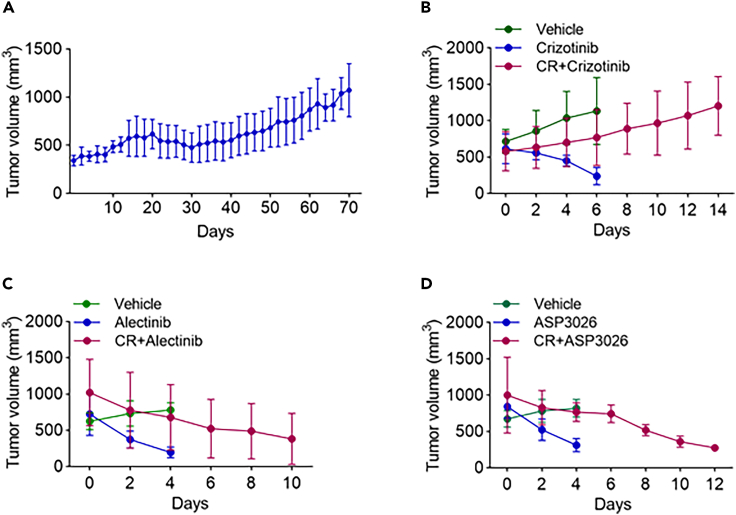
Anaplastic lymphoma kinase (ALK) inhibitor crizotinib has dramatic effect in non-small cell lung cancer patients with ALK rearrangement. However, most patients eventually develop resistance. To discover therapeutic targets to overcome crizotinib resistance (CR), we generated patient-derived xenograft CR mice and subjected them to phosphorylation profiling, together with CR mice treated with ASP3026 or alectinib. We identified 100 proteins with different phosphorylation status in CR mice. Among them, AXL phosphorylation was increased in CR mice, which could not be reversed by ASP3026 or alectinib. Importantly, the combined treatment of crizotinib and AXL inhibitor in CR mice significantly inhibited tumor growth, compared to crizotinib alone. We also found that SHC1 phosphorylation was increased in CR mice and SHC1 knockdown sensitized ALK-driven cells to crizotinib. Our study provides a new view of signaling pathways leading to CR, suggesting AXL and SHC1 as potential targets for combination therapy to overcome CR.
Keywords: Cancer; Drugs; Molecular biology.
© 2024 The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures





References
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
