

High resolution analysis of proteolytic substrate processing
- PMID: 39313096
- PMCID: PMC11513451
- DOI: 10.1016/j.jbc.2024.107812
High resolution analysis of proteolytic substrate processing
Abstract
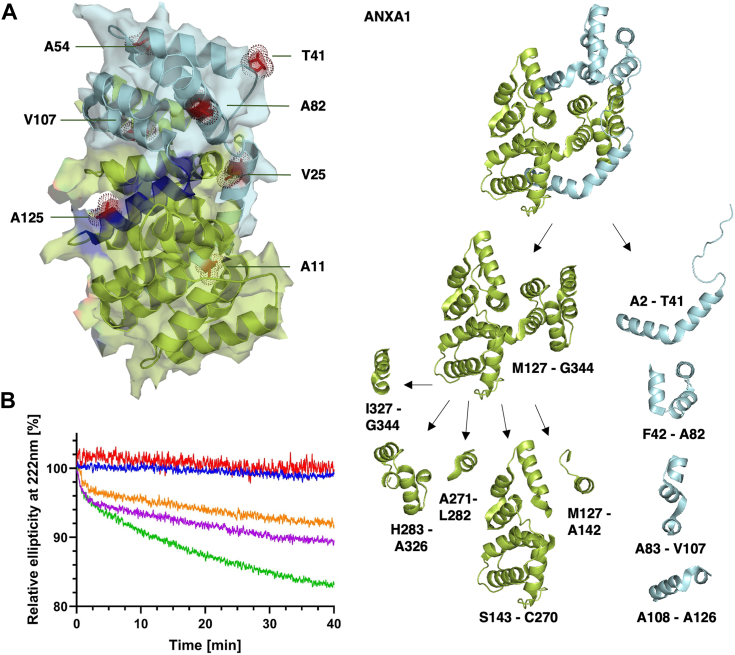
Members of the widely conserved high temperature requirement A (HtrA) family of serine proteases are involved in multiple aspects of protein quality control. In this context, they have been shown to efficiently degrade misfolded proteins or protein fragments. However, recent reports suggest that folded proteins can also be native substrates. To gain a deeper understanding of how folded proteins are initially processed and subsequently degraded into short peptides by human HTRA1, we established an integrated and quantitative approach using time-resolved mass spectrometry, CD spectroscopy, and bioinformatics. The resulting data provide high-resolution information on up to 178 individual proteolytic sites within folded ANXA1 (consisting of 346 amino acids), the relative frequency of cuts at each proteolytic site, the preferences of the protease for the amino acid sequence surrounding the scissile bond, as well as the degrees of sequential structural relaxation and unfolding of the substrate that occur during progressive degradation. Our workflow provides precise molecular insights into protease-substrate interactions, which could be readily adapted to address other posttranslational modifications such as phosphorylation in dynamic protein complexes.
Keywords: ANXA1; HTRA1; bioinformatics; protein degradation; protein processing; proteolysis; trypsin.
Copyright © 2024 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Conflict of interest The authors declare that they have no conflicts of interest with the contents of this article.
Figures






References
-
- Clausen T., Kaiser M., Huber R., Ehrmann M. HTRA proteases: regulated proteolysis in protein quality control. Nat. Rev. Mol. Cell Biol. 2011;12:152–162. - PubMed
-
- Clausen T., Southan C., Ehrmann M. The HtrA family of proteases. Implications for protein composition and cell fate. Mol. Cell. 2002;10:443–455. - PubMed
-
- Zurawa-Janicka D., Wenta T., Jarzab M., Skorko-Glonek J., Glaza P., Gieldon A., et al. Structural insights into the activation mechanisms of human HtrA serine proteases. Arch. Biochem. Biophys. 2017;621:6–23. - PubMed
-
- Eigenbrot C., Ultsch M., Lipari M.T., Moran P., Lin S.J., Ganesan R., et al. Structural and functional analysis of HtrA1 and its subdomains. Structure. 2012;20:1040–1050. - PubMed
-
- Truebestein L., Tennstaedt A., Monig T., Krojer T., Canellas F., Kaiser M., et al. Substrate-induced remodeling of the active site regulates human HTRA1 activity. Nat. Struct. Mol. Biol. 2011;18:386–388. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
