

Core and accessory genomic traits of Vibrio cholerae O1 drive lineage transmission and disease severity
- PMID: 39313510
- PMCID: PMC11420230
- DOI: 10.1038/s41467-024-52238-0
Core and accessory genomic traits of Vibrio cholerae O1 drive lineage transmission and disease severity
Abstract
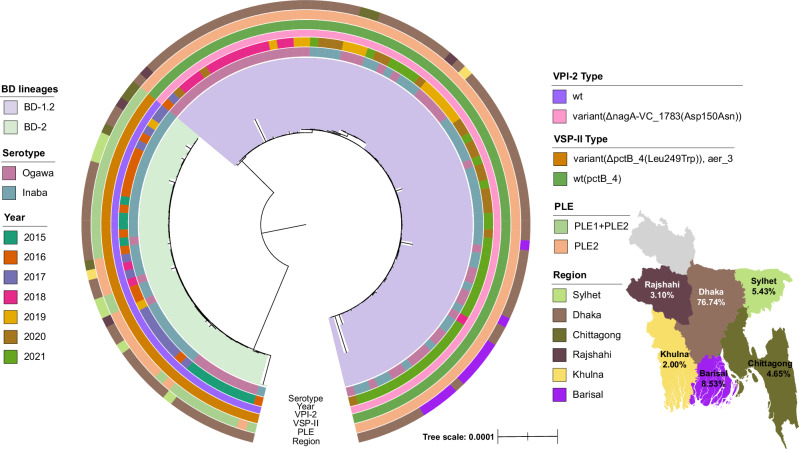
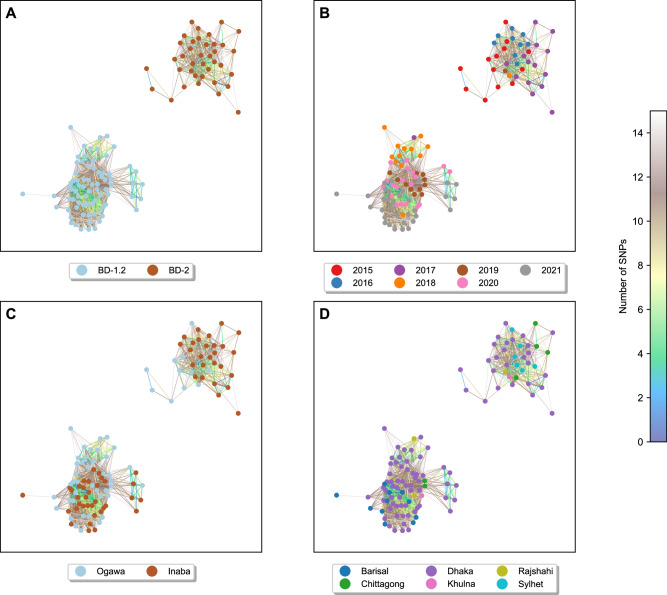
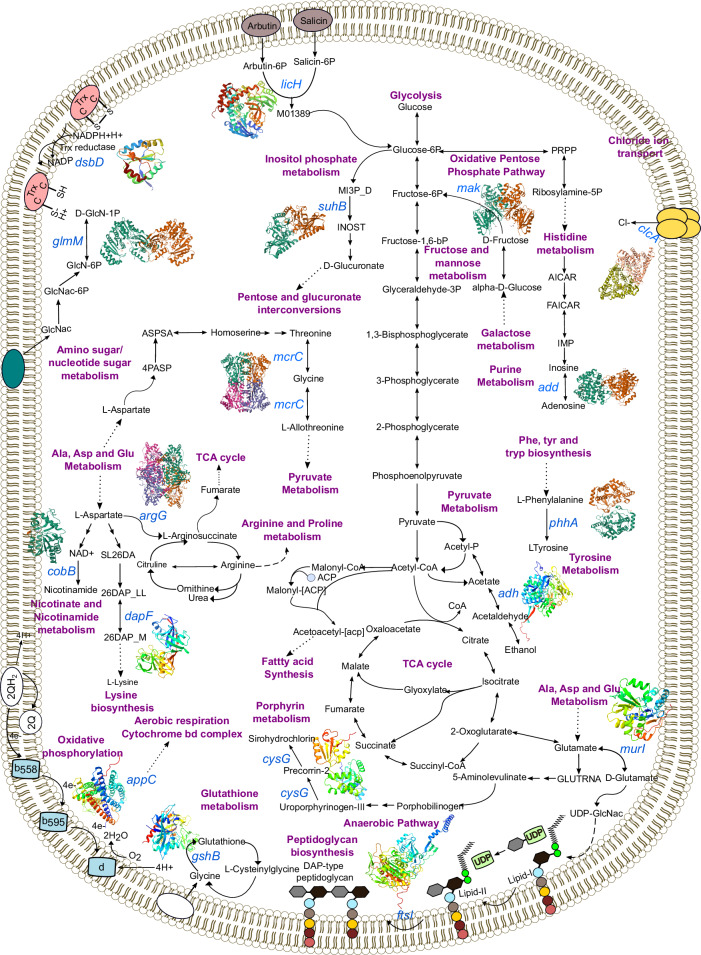
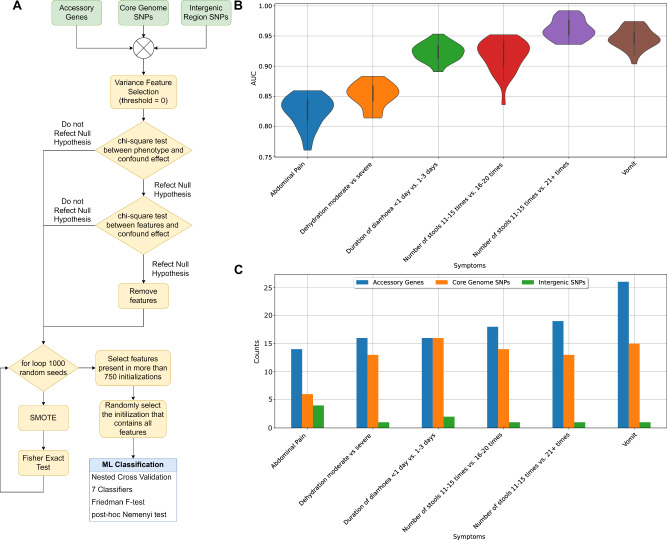
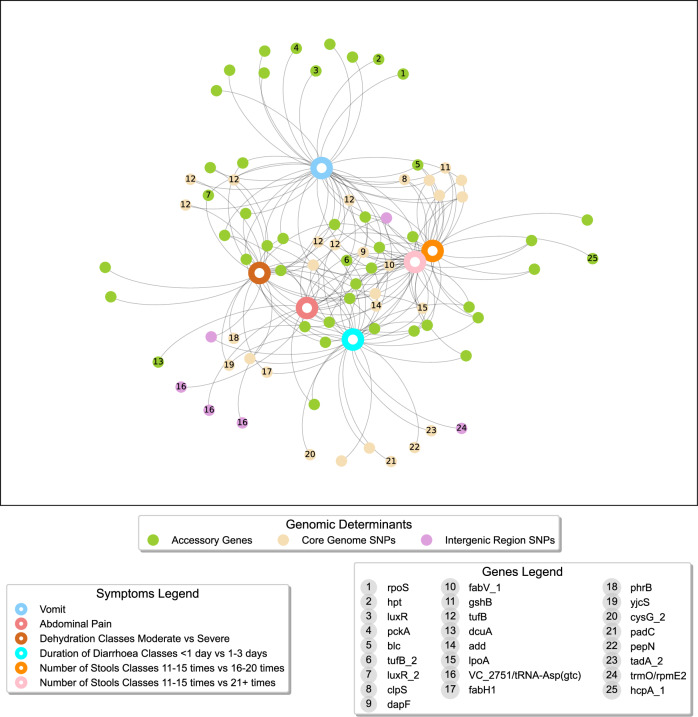
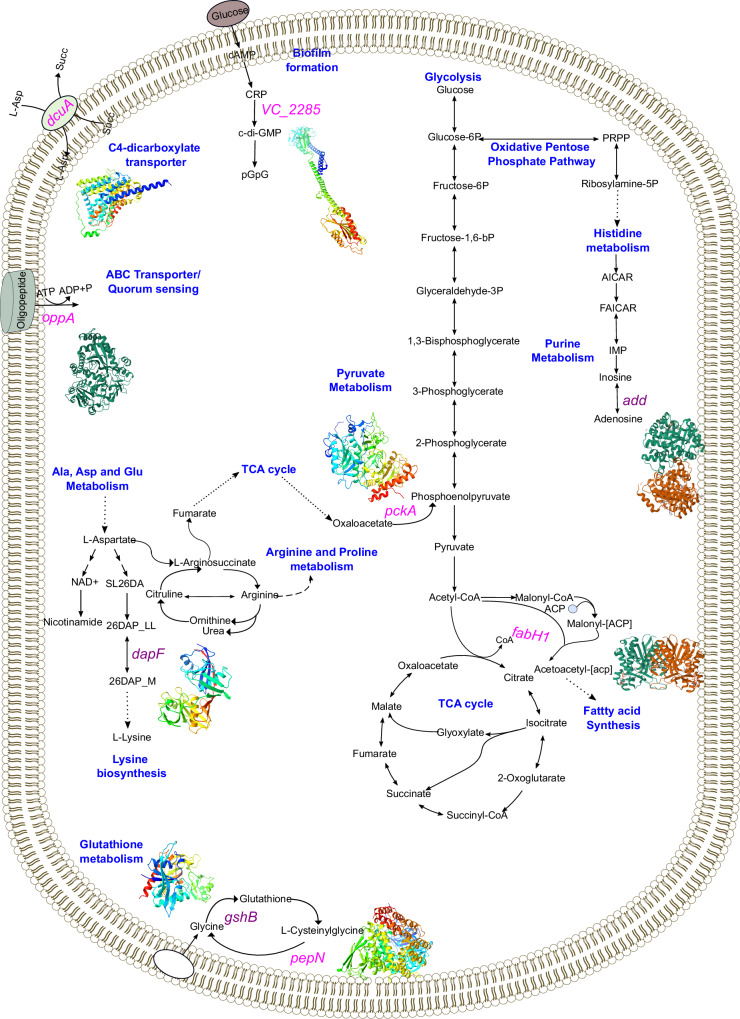
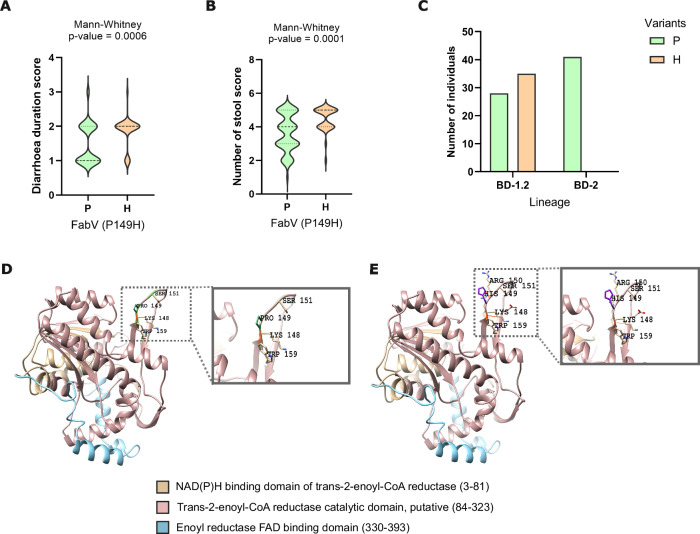
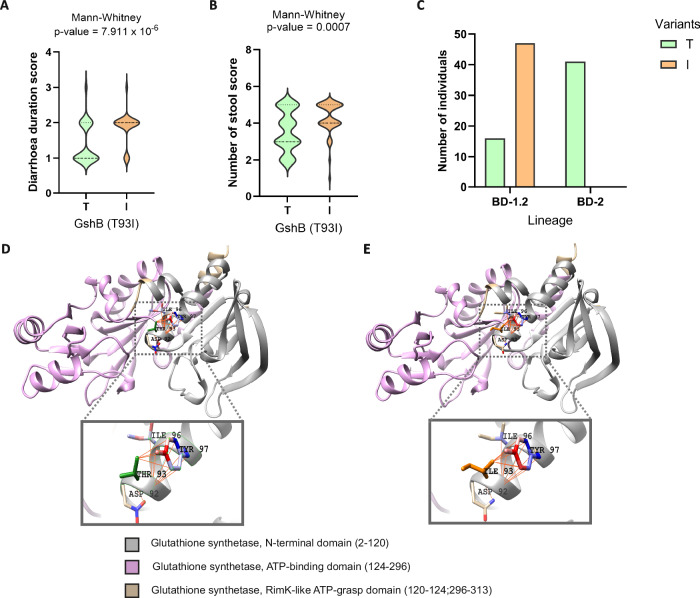
In Bangladesh, Vibrio cholerae lineages are undergoing genomic evolution, with increased virulence and spreading ability. However, our understanding of the genomic determinants influencing lineage transmission and disease severity remains incomplete. Here, we developed a computational framework using machine-learning, genome scale metabolic modelling (GSSM) and 3D structural analysis, to identify V. cholerae genomic traits linked to lineage transmission and disease severity. We analysed in-patients isolates from six Bangladeshi regions (2015-2021), and uncovered accessory genes and core SNPs unique to the most recent dominant lineage, with virulence, motility and bacteriophage resistance functions. We also found a strong correlation between V. cholerae genomic traits and disease severity, with some traits overlapping those driving lineage transmission. GSMM and 3D structure analysis unveiled a complex interplay between transcription regulation, protein interaction and stability, and metabolic networks, associated to lifestyle adaptation, intestinal colonization, acid tolerance and symptom severity. Our findings support advancing therapeutics and targeted interventions to mitigate cholera spread.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Banerjee, R., Das, B., Nair, G. B. & Basak, S. Dynamics in genome evolution of Vibrio cholerae. Infect. Genet. Evol.23, 32–41 (2014). - PubMed
Publication types
MeSH terms
Associated data
LinkOut - more resources
Full Text Sources
Medical
