

Regulation of intestinal senescence during cholestatic liver disease modulates barrier function and liver disease progression
- PMID: 39314550
- PMCID: PMC11418120
- DOI: 10.1016/j.jhepr.2024.101159
Regulation of intestinal senescence during cholestatic liver disease modulates barrier function and liver disease progression
Abstract
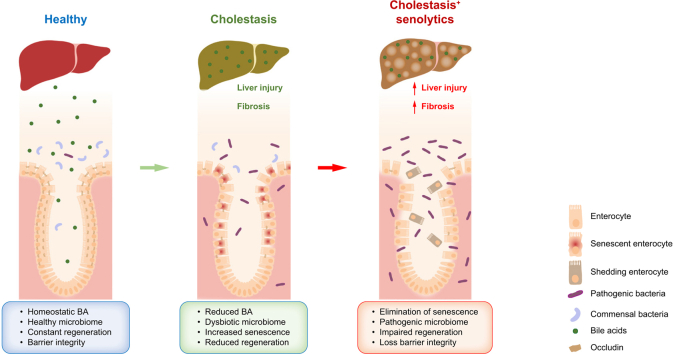
Background & aims: Senescence has been reported to have differential functions in cholangiocytes and hepatic stellate cells (HSCs) during human and murine cholestatic disease, being detrimental in biliary cells and anti-fibrotic in HSCs. Cholestatic liver disease is associated with loss of intestinal barrier function and changes in the microbiome, the mechanistic cause of which is undetermined.
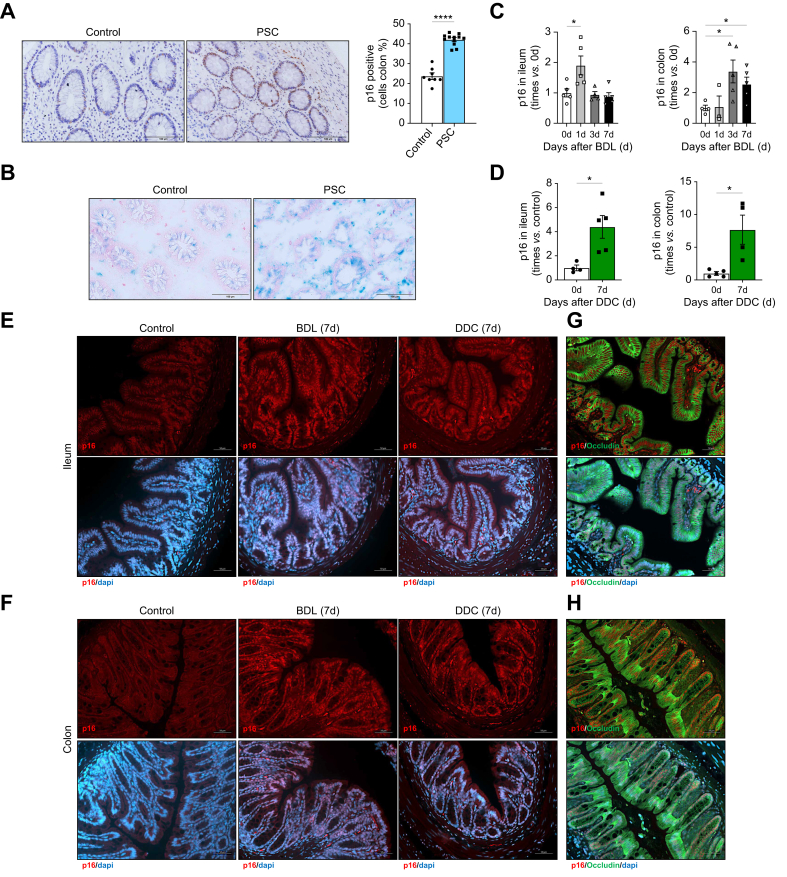
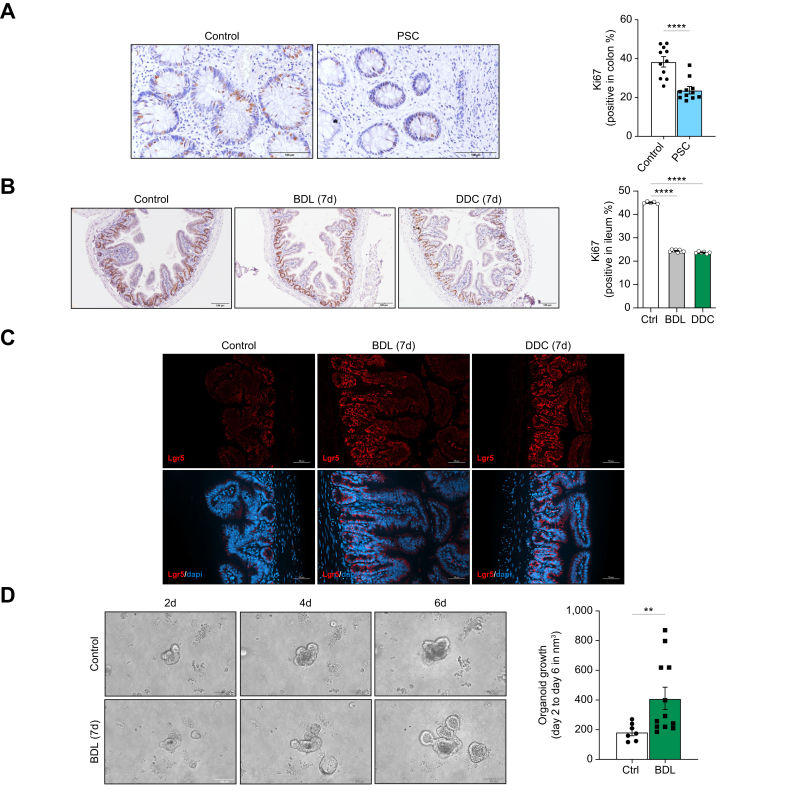
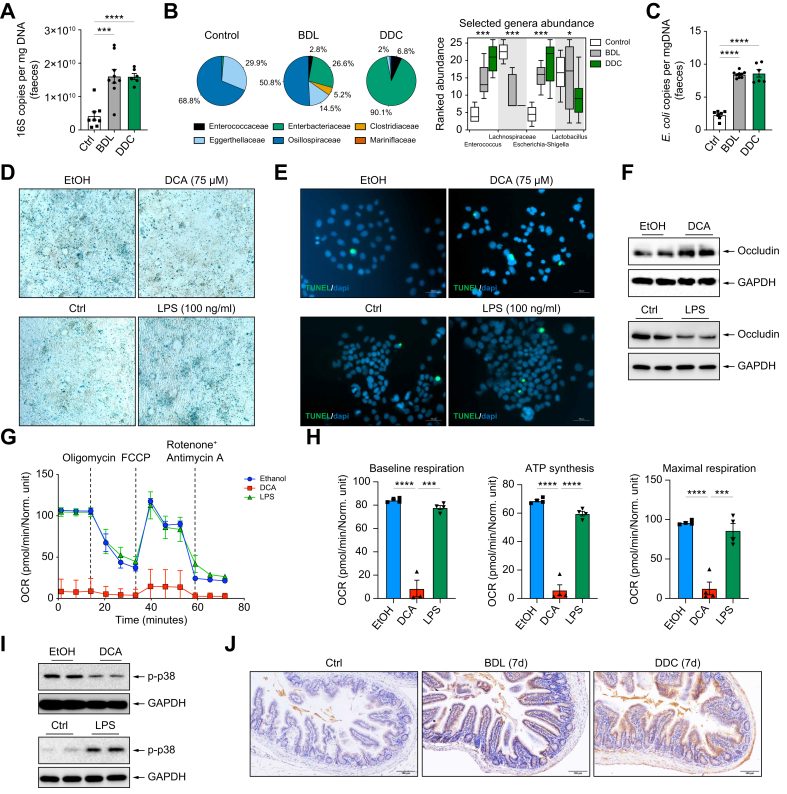
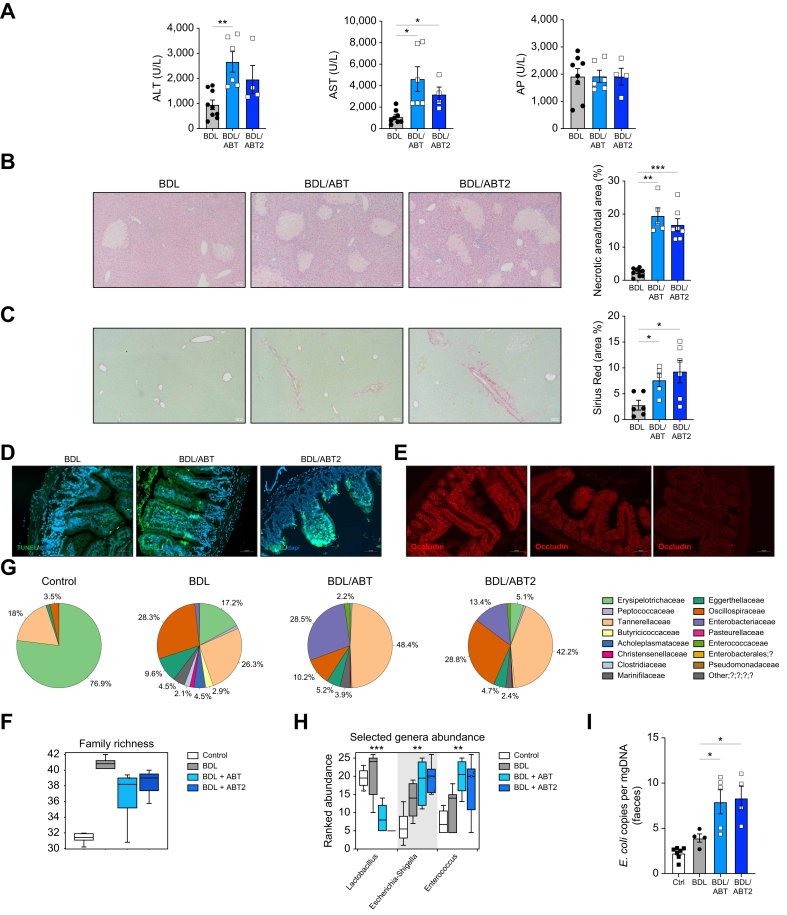
Methods: Intestinal samples were analysed from controls and patients with primary sclerosing cholangitis, as well as wild-type (WT) and p16-3MR transgenic mice. Cholestatic liver disease was induced by bile duct ligation (BDL) and DDC diet feeding. Fexaramine was used as an intestinal-restricted FXR agonist and antibiotics were given to eliminate the intestinal microbiome. Senescent cells were eliminated in p16-3MR mice with ganciclovir and in WT mice with the senolytic drug ABT-263. In vitro studies were done in intestinal CaCo-2 cells and organoids were generated from intestinal crypts isolated from mice.
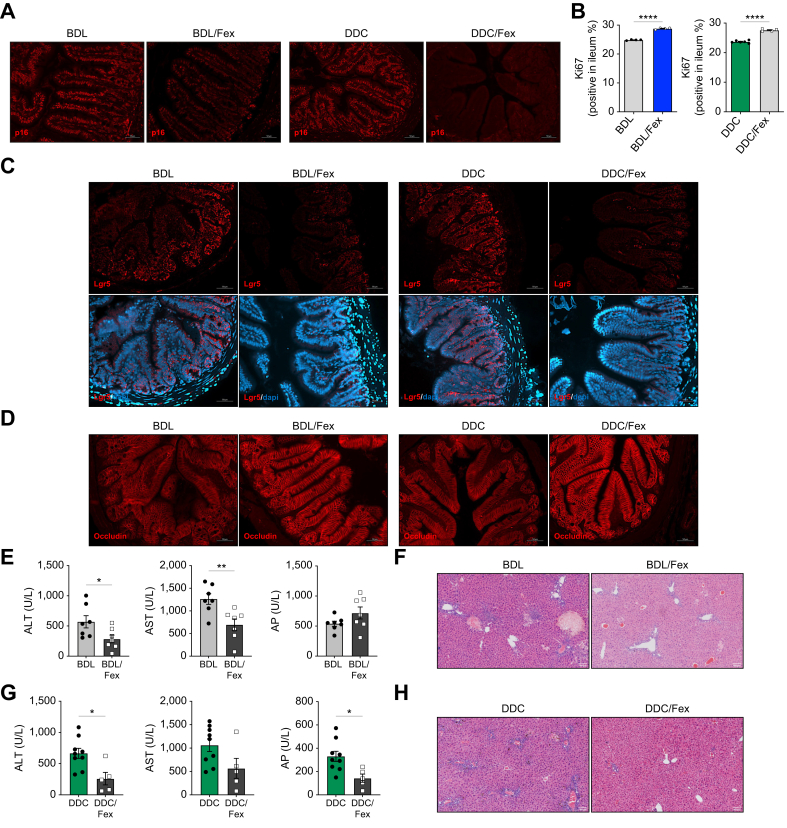
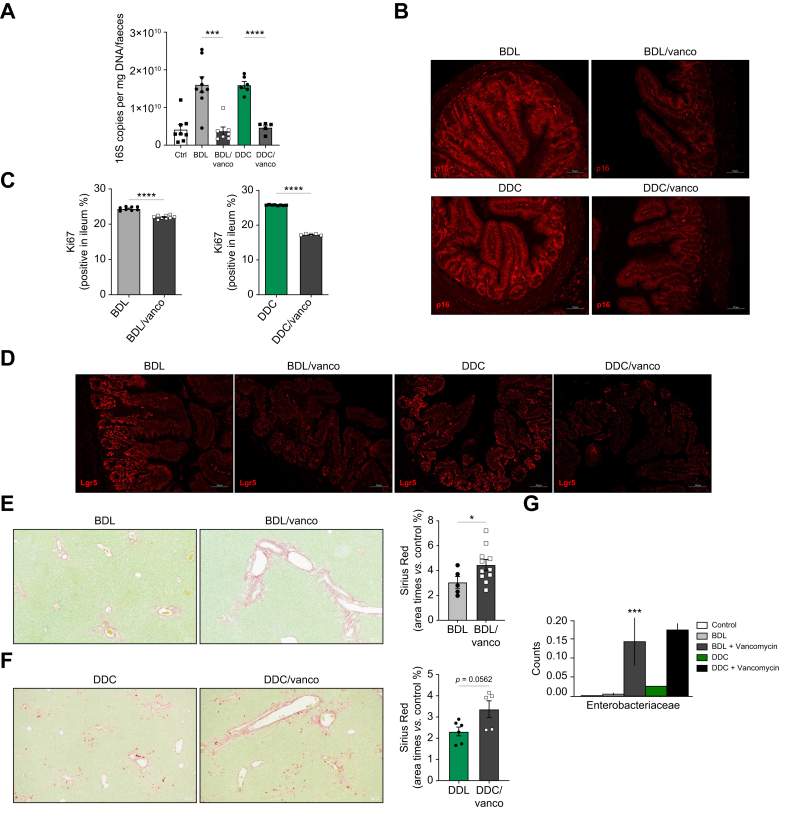
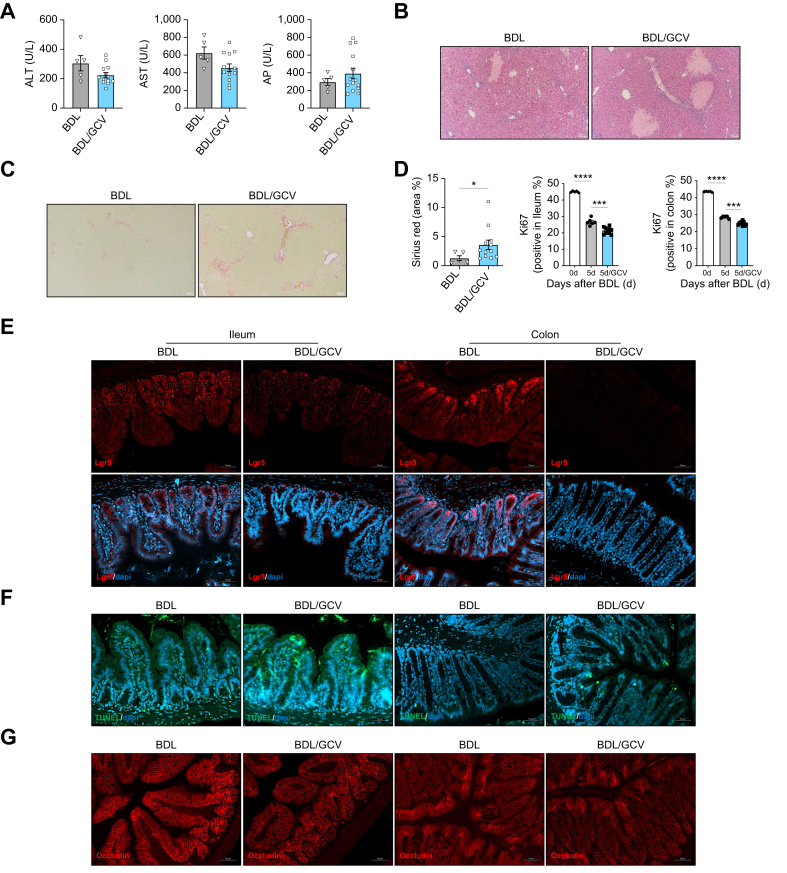
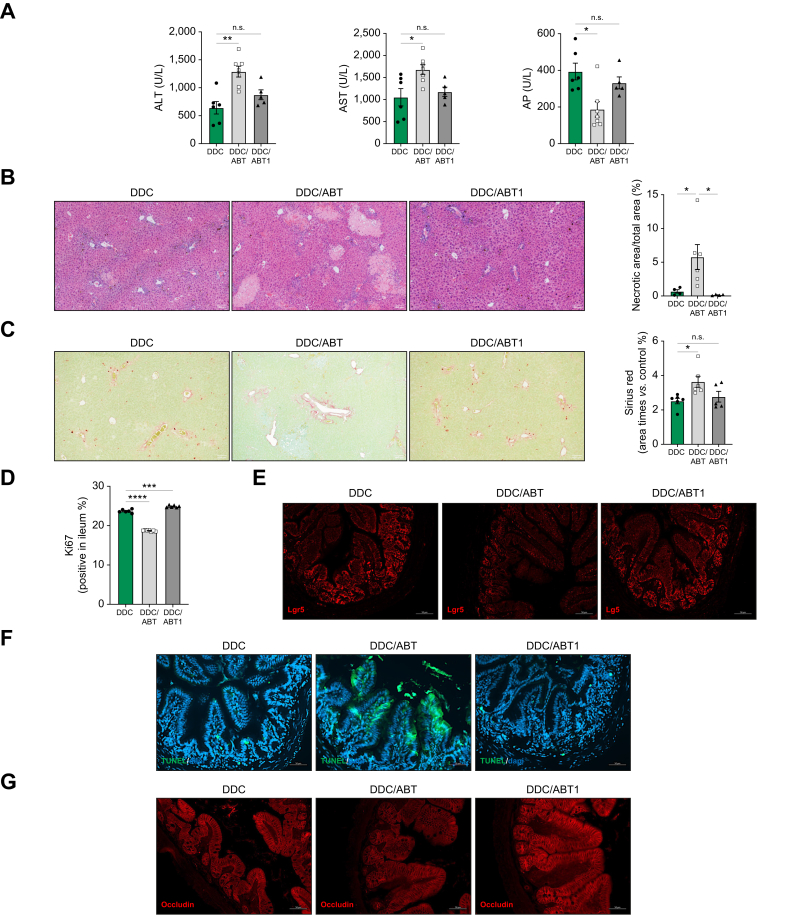
Results: Herein, we show increased senescence in intestinal epithelial cells (IECs) in patients with primary sclerosing cholangitis and in mice after BDL and DDC diet feeding. Intestinal senescence was increased in response to reduced exposure to bile acids and increased presence of lipopolysaccharide in vitro and in vivo during cholestatic liver disease. Senescence of IECs was associated with lower proliferation but increased intestinal stem cell activation, as supported by increased organoid growth from intestinal stem cells. Elimination of senescent cells with genetic and pharmacological approaches exacerbated liver injury and fibrosis during cholestatic liver disease, which was associated with increased IEC apoptosis and permeability.
Conclusions: Senescence occurs in IECs during cholestatic disease and the elimination of senescent cells has a detrimental impact on the gut-liver axis. Our results point to cell-specific rather than systemic targeting of senescence as a therapeutic approach to treat cholestatic liver disease.
Impact and implications: Cholestatic liver disease associates with the dysregulation of intestinal barrier function, while the mechanisms mediating the disruption of the gut-liver axis remain largely undefined. Here, we demonstrate that senescence, a cellular response to stress, is activated in intestinal cells during cholestatic liver disease in humans and mice. Mechanistically, we demonstrate that the reduction of bile acids and the increased presence of bacterial products mediate the activation of intestinal senescence during cholestatic liver disease. Importantly, the elimination of these senescent cells promotes further damage to the intestine that aggravates liver disease, with increased tissue damage and fibrosis. Our results provide evidence that therapeutic strategies to treat cholestatic liver disease by eliminating senescent cells may have unwanted effects in the intestine and support the need to develop cell/organ-specific approaches.
Keywords: Senescence; cholestasis; intestine; liver; senolytics.
© 2024 The Authors.
Figures
References
-
- Campisi J., d'Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. 2007;8:729–740. - PubMed
-
- Munoz-Espin D., Serrano M. Cellular senescence: from physiology to pathology. Nat Rev Mol Cell Biol. 2014;15:482–496. - PubMed
-
- Sasaki M., Ikeda H., Haga H., et al. Frequent cellular senescence in small bile ducts in primary biliary cirrhosis: a possible role in bile duct loss. J Pathol. 2005;205:451–459. - PubMed
LinkOut - more resources
Full Text Sources
