

p53-dependent crosstalk between DNA replication integrity and redox metabolism mediated through a NRF2-PARP1 axis
- PMID: 39315696
- PMCID: PMC11551750
- DOI: 10.1093/nar/gkae811
p53-dependent crosstalk between DNA replication integrity and redox metabolism mediated through a NRF2-PARP1 axis
Abstract
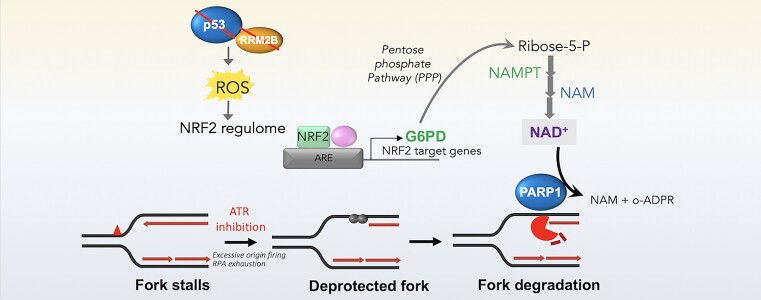
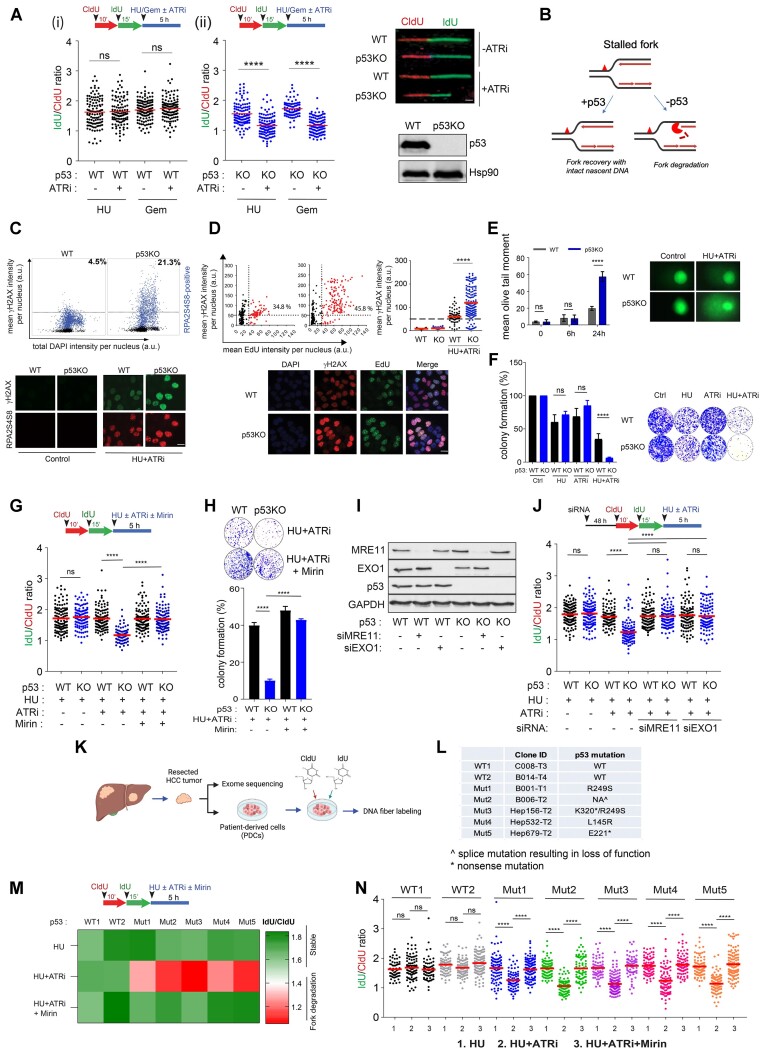
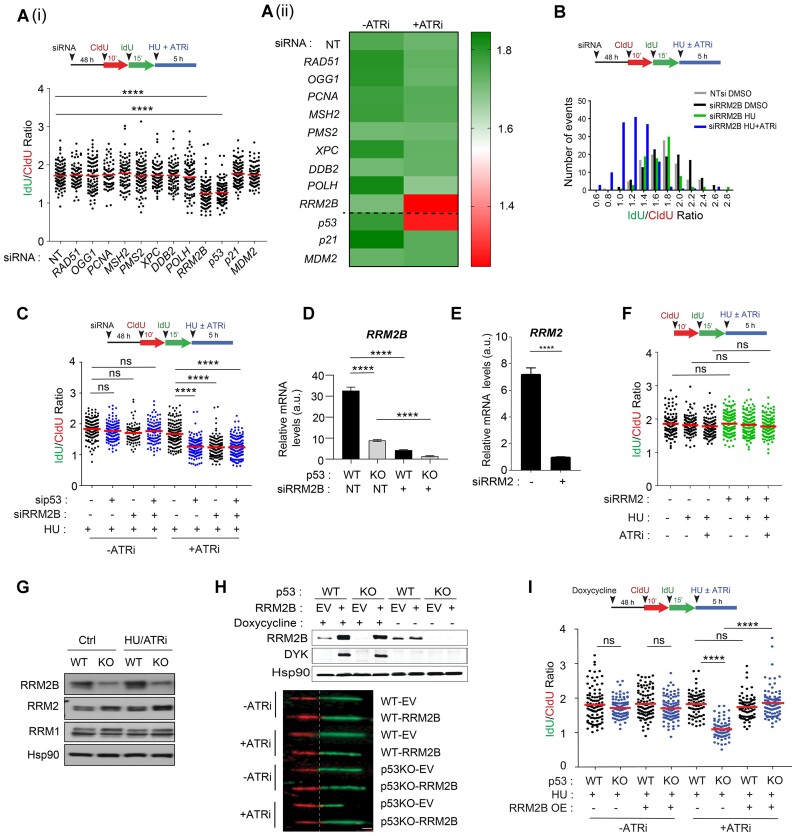
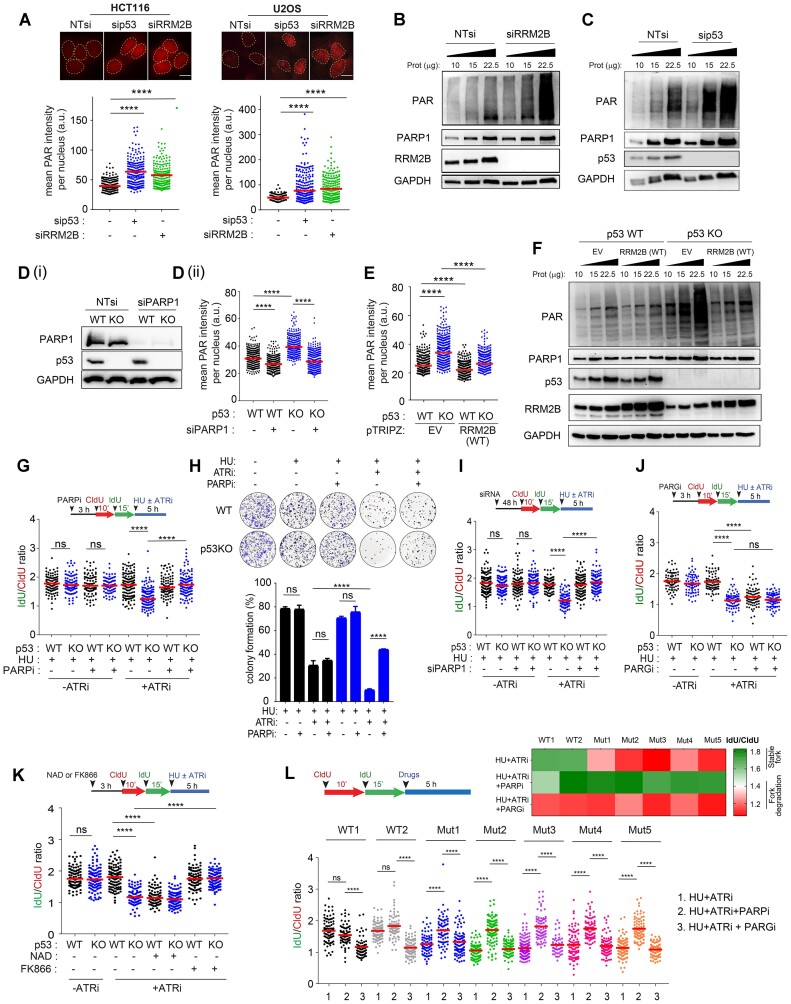
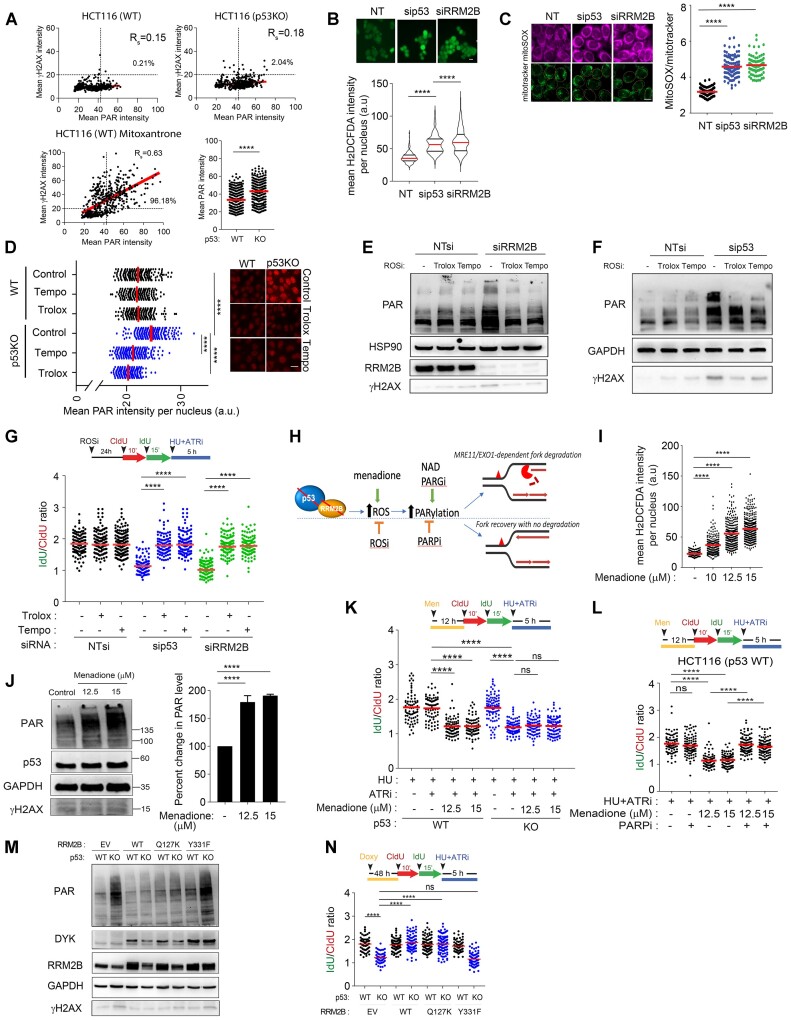
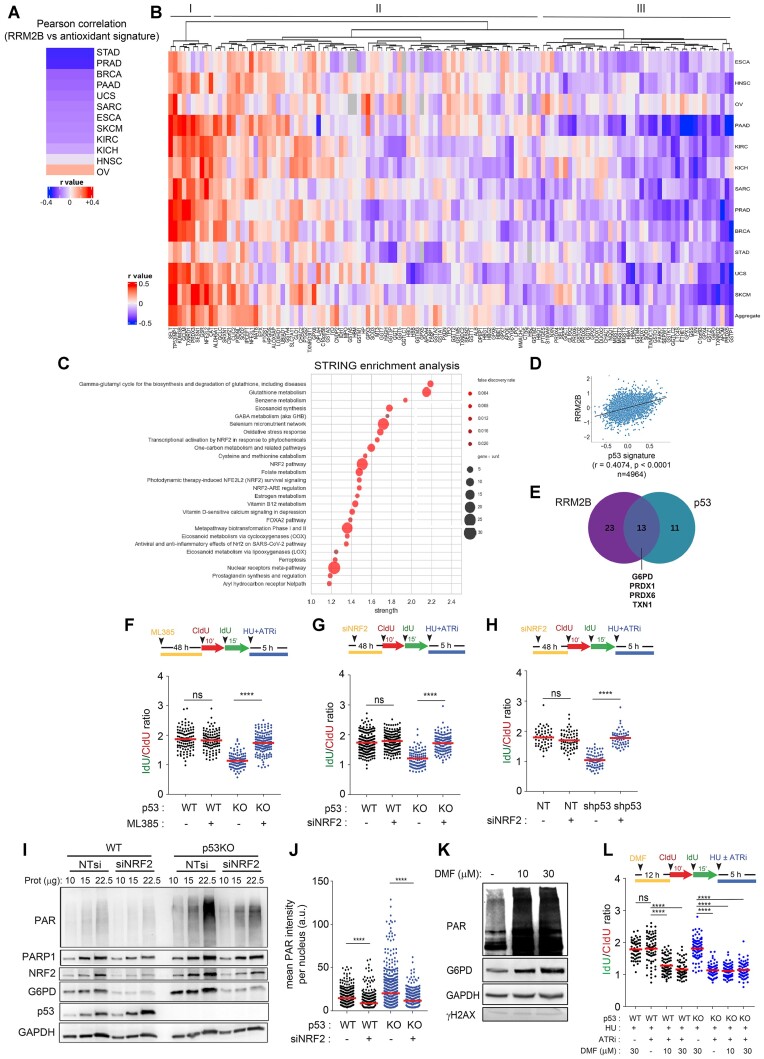
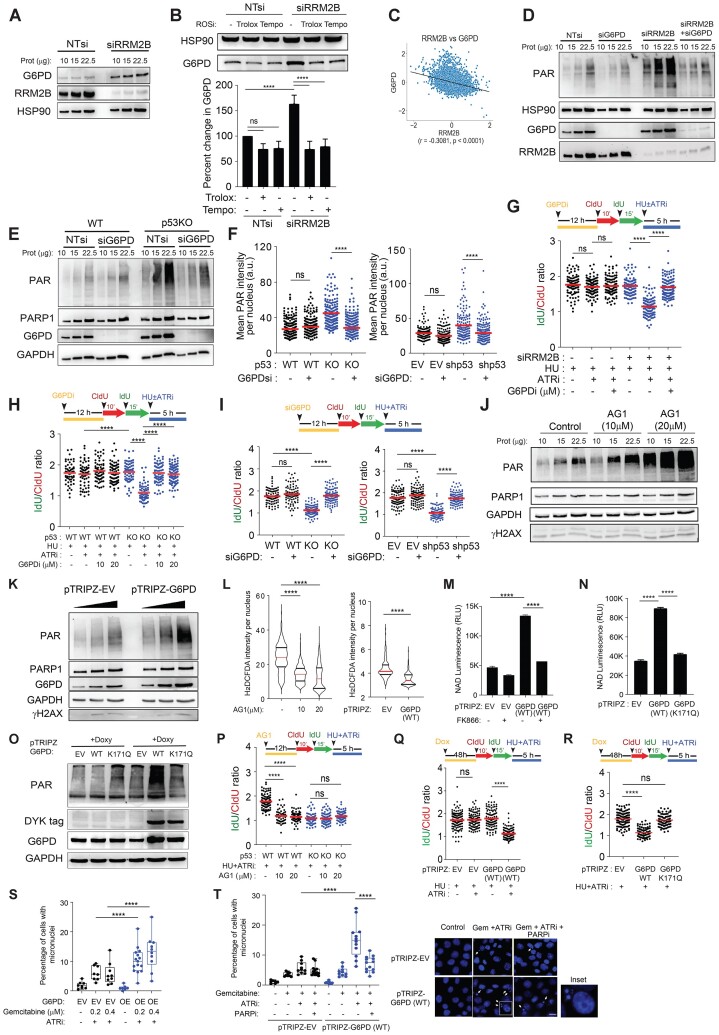
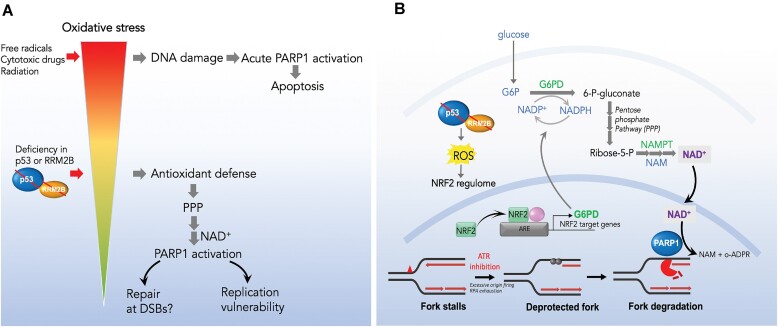
Mechanisms underlying p53-mediated protection of the replicating genome remain elusive, despite the quintessential role of p53 in maintaining genomic stability. Here, we uncover an unexpected function of p53 in curbing replication stress by limiting PARP1 activity and preventing the unscheduled degradation of deprotected stalled forks. We searched for p53-dependent factors and elucidated RRM2B as a prime factor. Deficiency in p53/RRM2B results in the activation of an NRF2 antioxidant transcriptional program, with a concomitant elevation in basal PARylation in cells. Dissecting the consequences of p53/RRM2B loss revealed a crosstalk between redox metabolism and genome integrity that is negotiated through a hitherto undescribed NRF2-PARP1 axis, and pinpoint G6PD as a primary oxidative stress-induced NRF2 target and activator of basal PARylation. This study elucidates how loss of p53 could be destabilizing for the replicating genome and, importantly, describes an unanticipated crosstalk between redox metabolism, PARP1 and p53 tumor suppressor pathway that is broadly relevant in cancers and can be leveraged therapeutically.
© The Author(s) 2024. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures
References
-
- Berti M., Cortez D., Lopes M.. The plasticity of DNA replication forks in response to clinically relevant genotoxic stress. Nat. Rev. Mol. Cell Biol. 2020; 21:633–651. - PubMed
-
- Casper A.M., Nghiem P., Arlt M.F., Glover T.W.. ATR regulates fragile site stability. Cell. 2002; 111:779–789. - PubMed
-
- Cortez D., Guntuku S., Qin J., Elledge S.J.. ATR and ATRIP: partners in checkpoint signaling. Science. 2001; 294:1713–1716. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
