

The effect of traumatic brain injury on learning and memory: A synaptic focus
- PMID: 39316552
- PMCID: PMC11909778
- DOI: 10.1177/10738584241275583
The effect of traumatic brain injury on learning and memory: A synaptic focus
Abstract
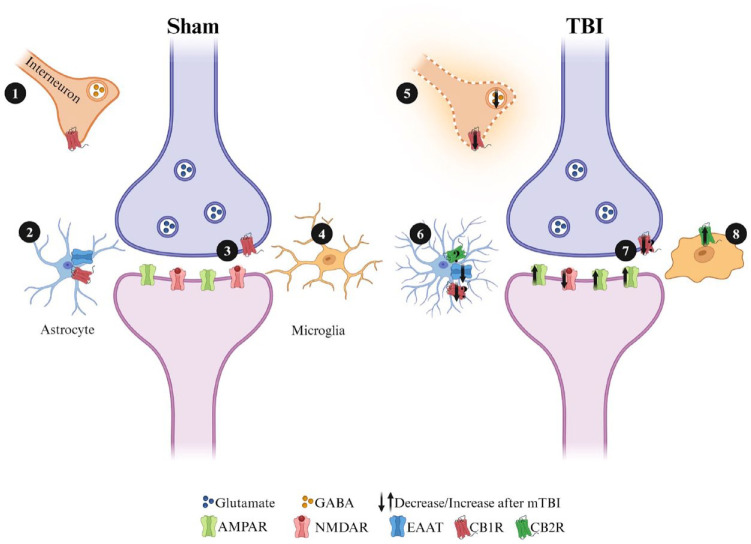
Deficits in learning and memory are some of the most commonly reported symptoms following a traumatic brain injury (TBI). We will examine whether the neural basis of these deficits stems from alterations to bidirectional synaptic plasticity within the hippocampus. Although the CA1 subregion of the hippocampus has been a focus of TBI research, the dentate gyrus should also be given attention as it exhibits a unique ability for adult neurogenesis, a process highly susceptible to TBI-induced damage. This review examines our current understanding of how TBI results in deficits in synaptic plasticity, as well as how TBI-induced changes in endocannabinoid (eCB) systems may drive these changes. Through the synthesis and amalgamation of existing data, we propose a possible mechanism for eCB-mediated recovery in synaptic plasticity deficits. This hypothesis is based on the plausible roles of CB1 receptors in regulating inhibitory tone, influencing astrocytes and microglia, and modulating glutamate release. Dysregulation of the eCBs may be responsible for deficits in synaptic plasticity and learning following TBI. Taken together, the existing evidence indicates eCBs may contribute to TBI manifestation, pathogenesis, and recovery, but it also suggests there may be a therapeutic role for the eCB system in TBI.
Keywords: dentate gyrus; electrophysiology; endocannabinoids; hippocampus; long-term depression; long-term potentiation; synaptic plasticity.
Conflict of interest statement
Declaration of Conflicting InterestsThe author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Figures

Similar articles
-
Traumatic brain injury modifies synaptic plasticity in newly-generated granule cells of the adult hippocampus.Exp Neurol. 2021 Feb;336:113527. doi: 10.1016/j.expneurol.2020.113527. Epub 2020 Nov 11. Exp Neurol. 2021. PMID: 33188818 Free PMC article.
-
Prenatal Ethanol Exposure Persistently Alters Endocannabinoid Signaling and Endocannabinoid-Mediated Excitatory Synaptic Plasticity in Ventral Tegmental Area Dopamine Neurons.J Neurosci. 2017 Jun 14;37(24):5798-5808. doi: 10.1523/JNEUROSCI.3894-16.2017. Epub 2017 May 5. J Neurosci. 2017. PMID: 28476947 Free PMC article.
-
Endocannabinoid-mediated synaptic plasticity in the CNS.Annu Rev Neurosci. 2006;29:37-76. doi: 10.1146/annurev.neuro.29.051605.112834. Annu Rev Neurosci. 2006. PMID: 16776579 Review.
-
Endocannabinoid signaling and long-term synaptic plasticity.Annu Rev Physiol. 2009;71:283-306. doi: 10.1146/annurev.physiol.010908.163149. Annu Rev Physiol. 2009. PMID: 19575681 Free PMC article. Review.
-
Glucocorticoid Receptor Overexpression in the Dorsal Hippocampus Attenuates Spatial Learning and Synaptic Plasticity Deficits after Pediatric Traumatic Brain Injury.J Neurotrauma. 2022 Jul;39(13-14):979-998. doi: 10.1089/neu.2022.0012. J Neurotrauma. 2022. PMID: 35293260 Free PMC article.
Cited by
-
Panaroma of microglia in traumatic brain injury: a bibliometric analysis and visualization study during 2000-2023.Front Cell Neurosci. 2024 Nov 7;18:1495542. doi: 10.3389/fncel.2024.1495542. eCollection 2024. Front Cell Neurosci. 2024. PMID: 39575155 Free PMC article.
References
-
- Albensi BC, Sullivan PG, Thompson MB, Scheff SW, Mattson MP. 2000. Cyclosporin ameliorates traumatic brain-injury-induced alterations of hippocampal synaptic plasticity. Exp Neurol 162(2):385–9. - PubMed
-
- Alkadhi KA. 2019. Cellular and molecular differences between area CA1 and the dentate gyrus of the hippocampus. Mol Neurobiol 56(9):6566–80. - PubMed
-
- Almeida-Suhett CP, Prager EM, Pidoplichko V, Figueiredo TH, Marini AM, and others. 2015. GABAergic interneuronal loss and reduced inhibitory synaptic transmission in the hippocampal CA1 region after mild traumatic brain injury. Exp Neurol 273:11–23. - PubMed
-
- Amenta PS, Jallo JI, Tuma RF, Elliott MB. 2012. A cannabinoid type 2 receptor agonist attenuates blood–brain barrier damage and neurodegeneration in a murine model of traumatic brain injury. J Neurosci Res 90(12):2293–305. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
