

Single-cell m6A profiling in the mouse brain uncovers cell type-specific RNA methylomes and age-dependent differential methylation
- PMID: 39317796
- PMCID: PMC11614689
- DOI: 10.1038/s41593-024-01768-3
Single-cell m6A profiling in the mouse brain uncovers cell type-specific RNA methylomes and age-dependent differential methylation
Abstract
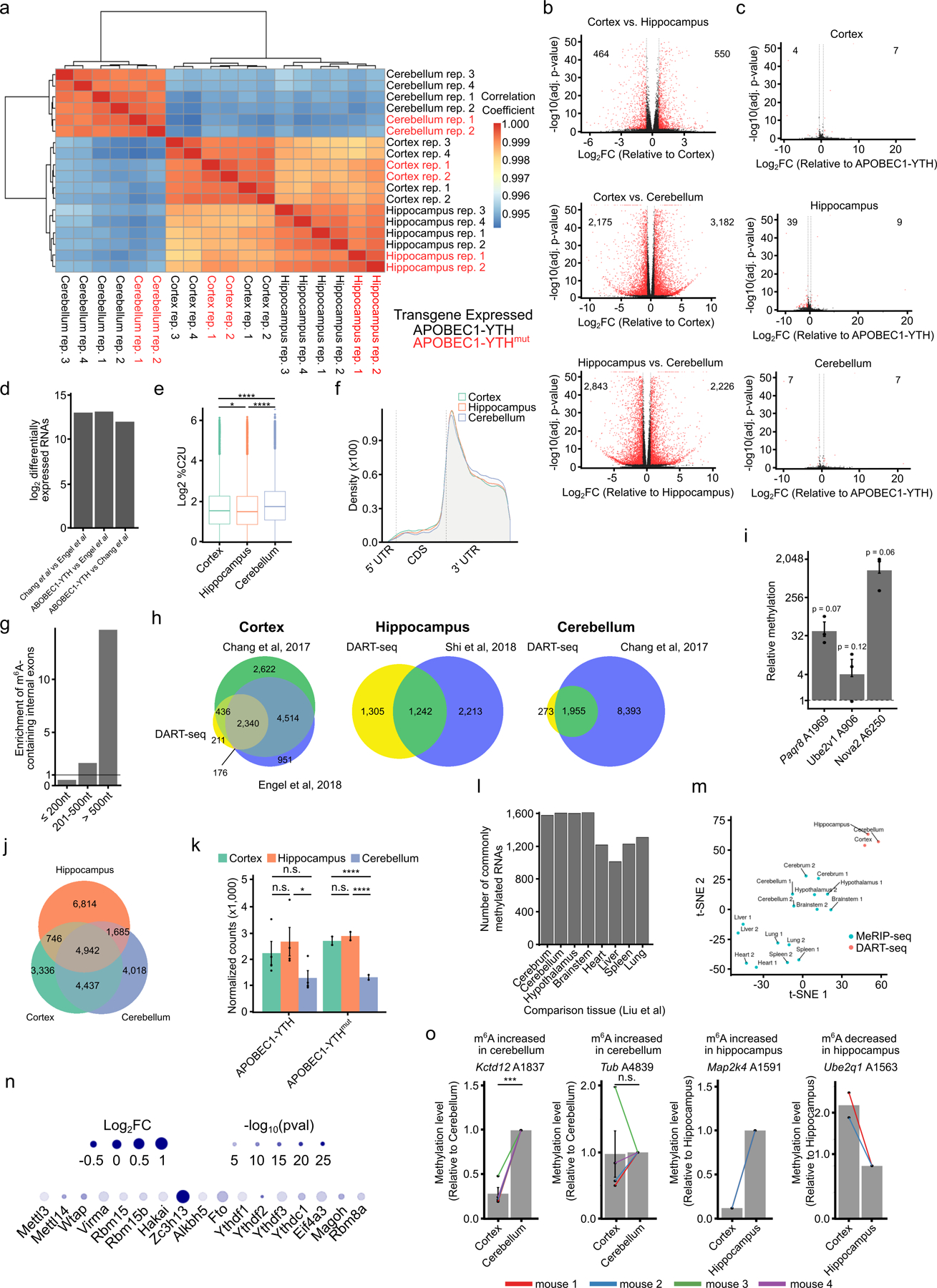
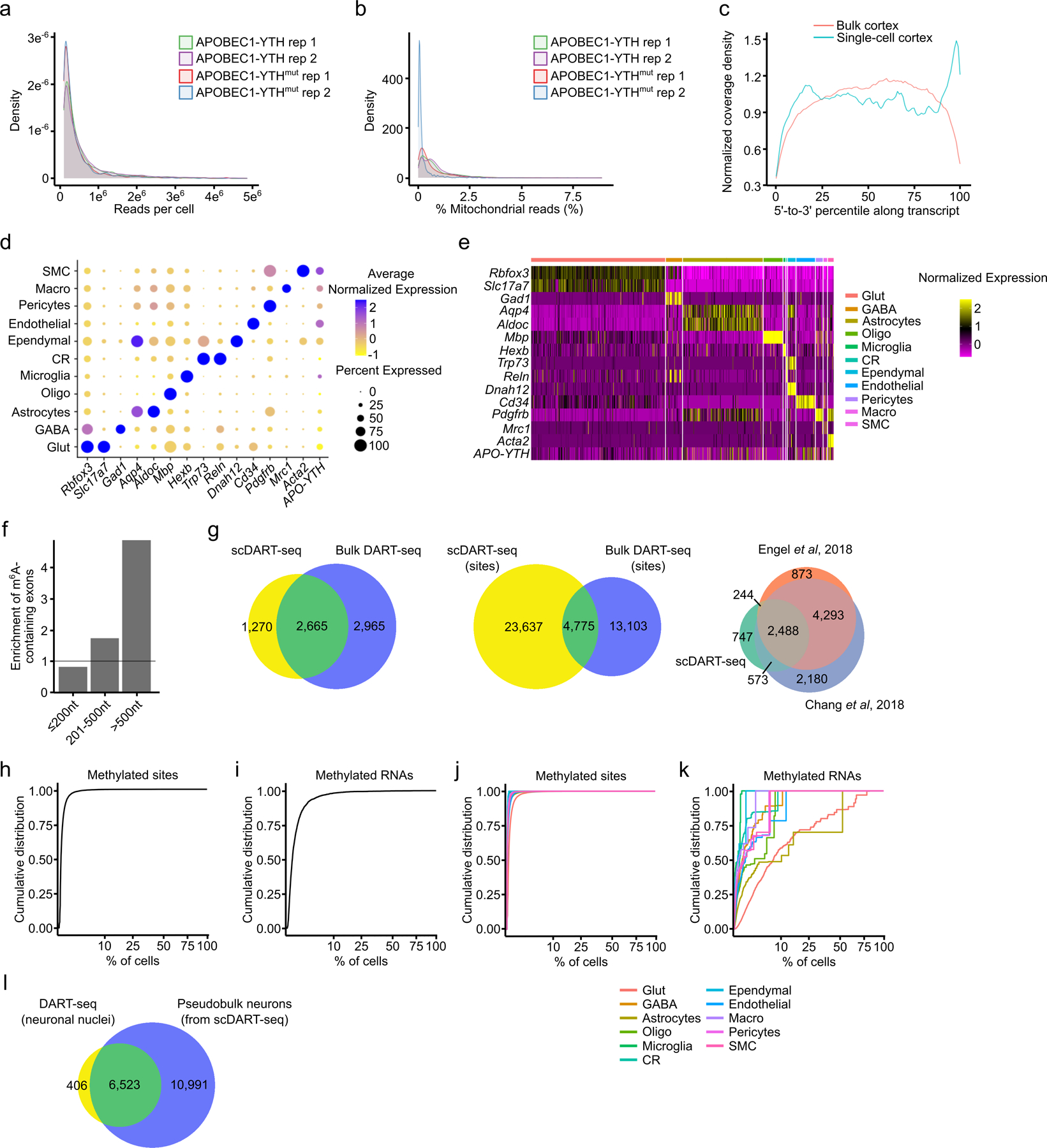
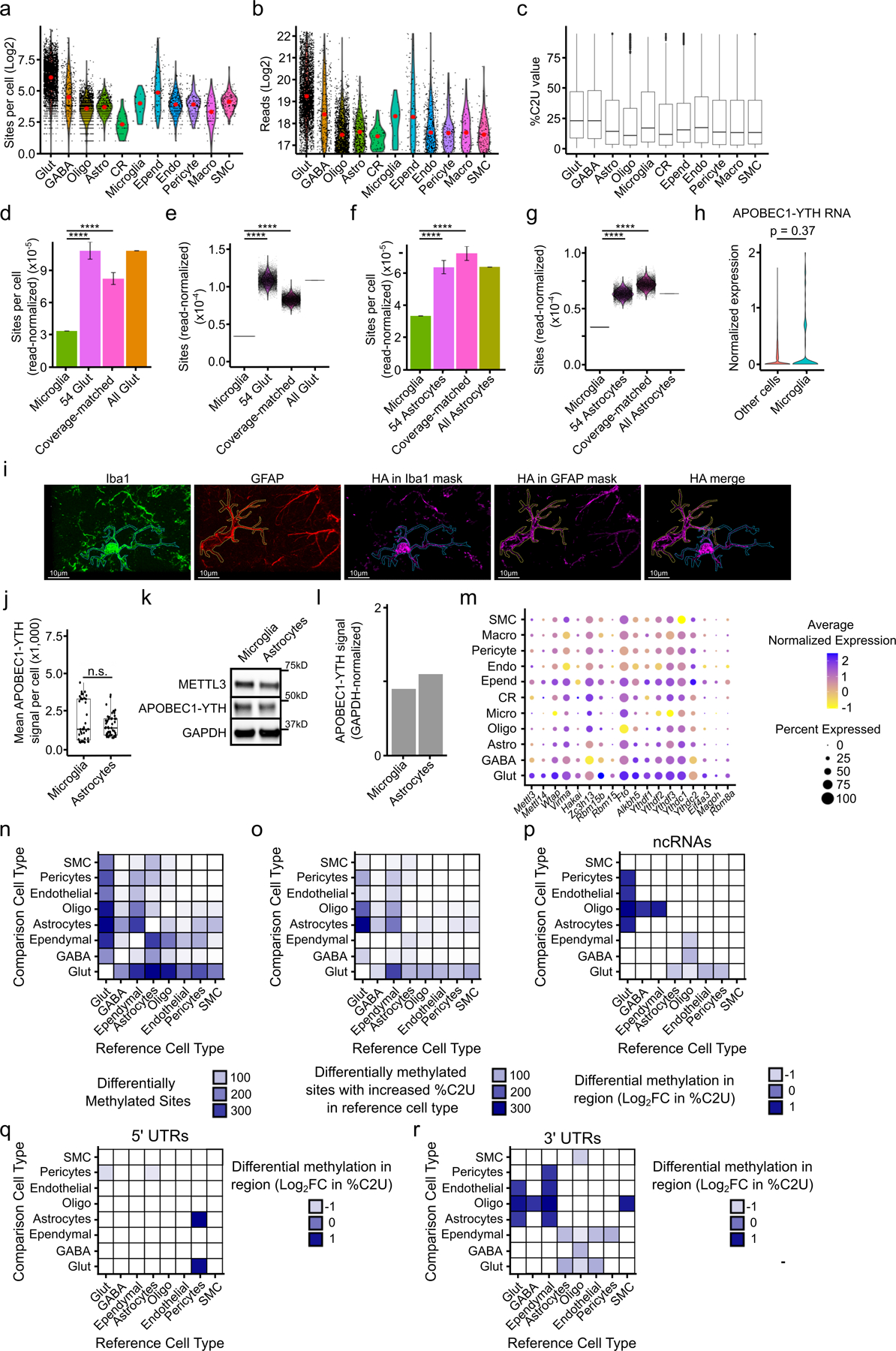
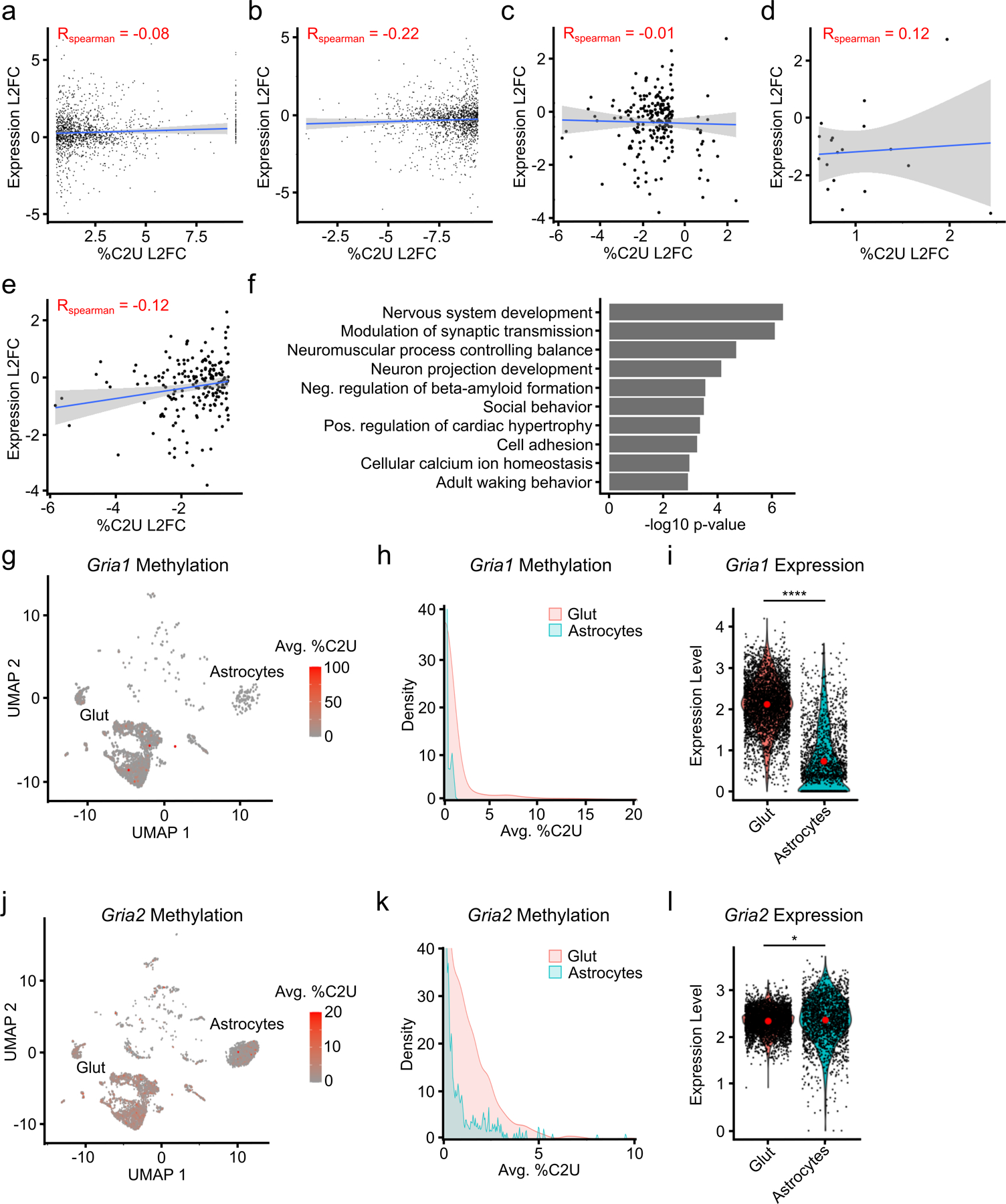
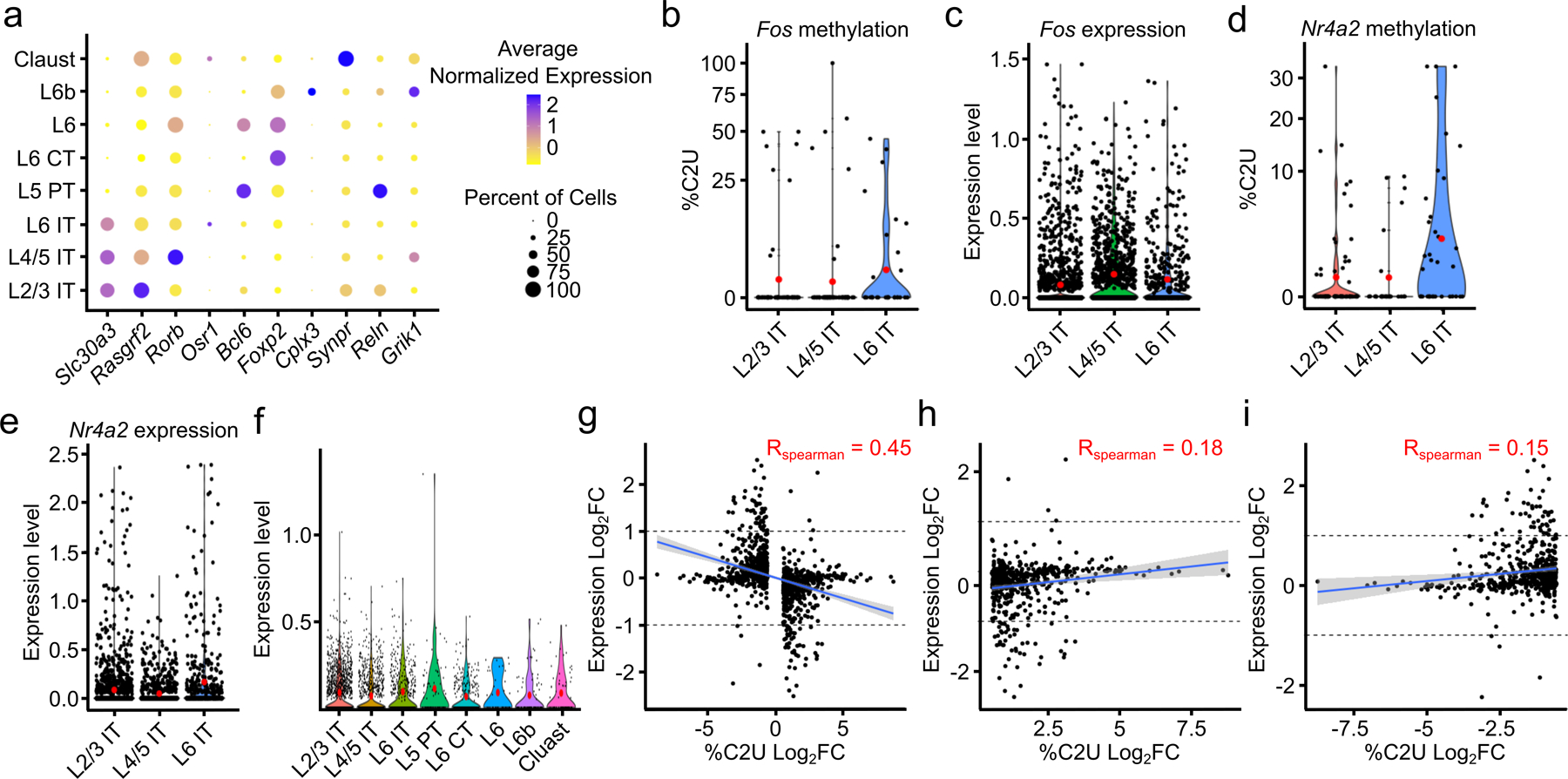
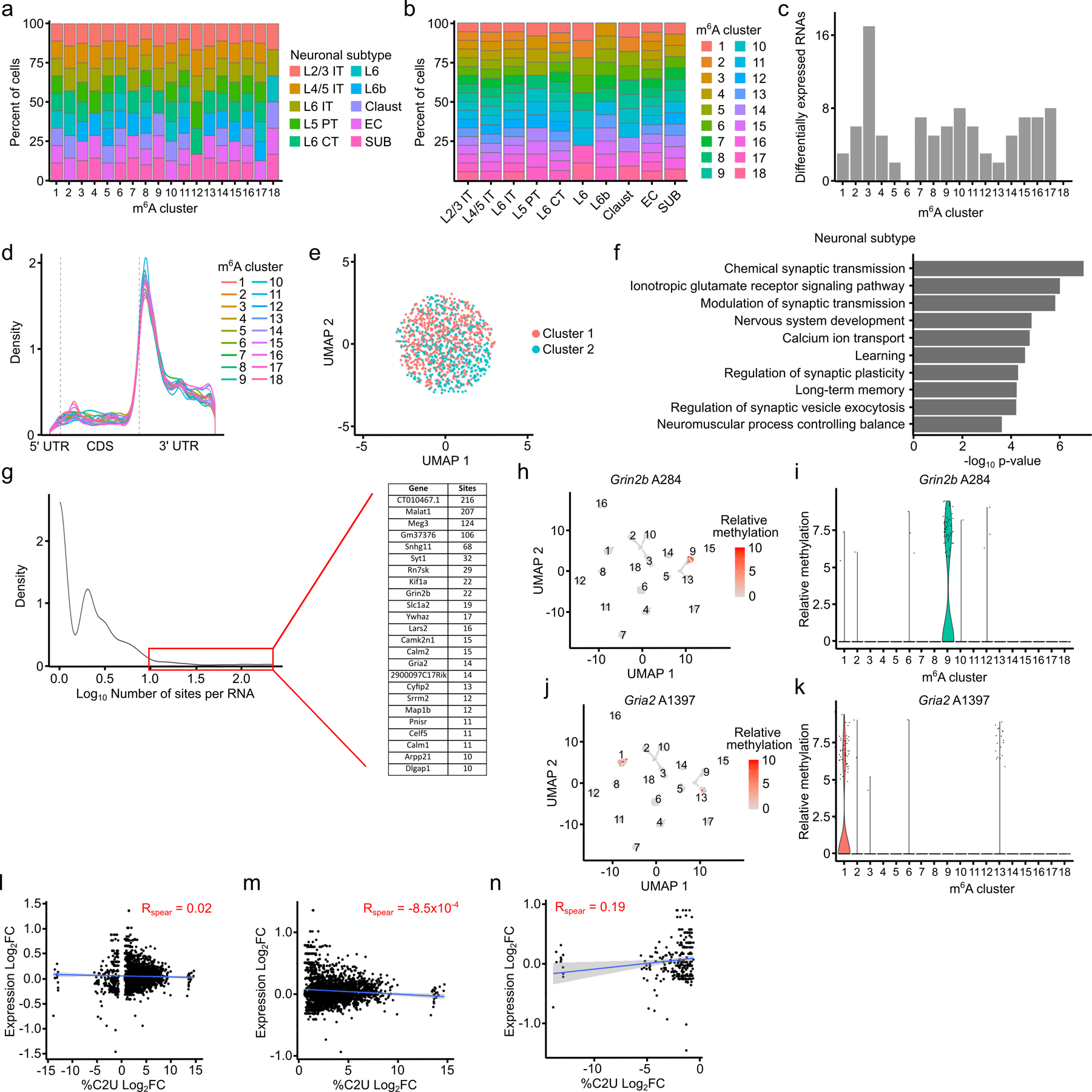
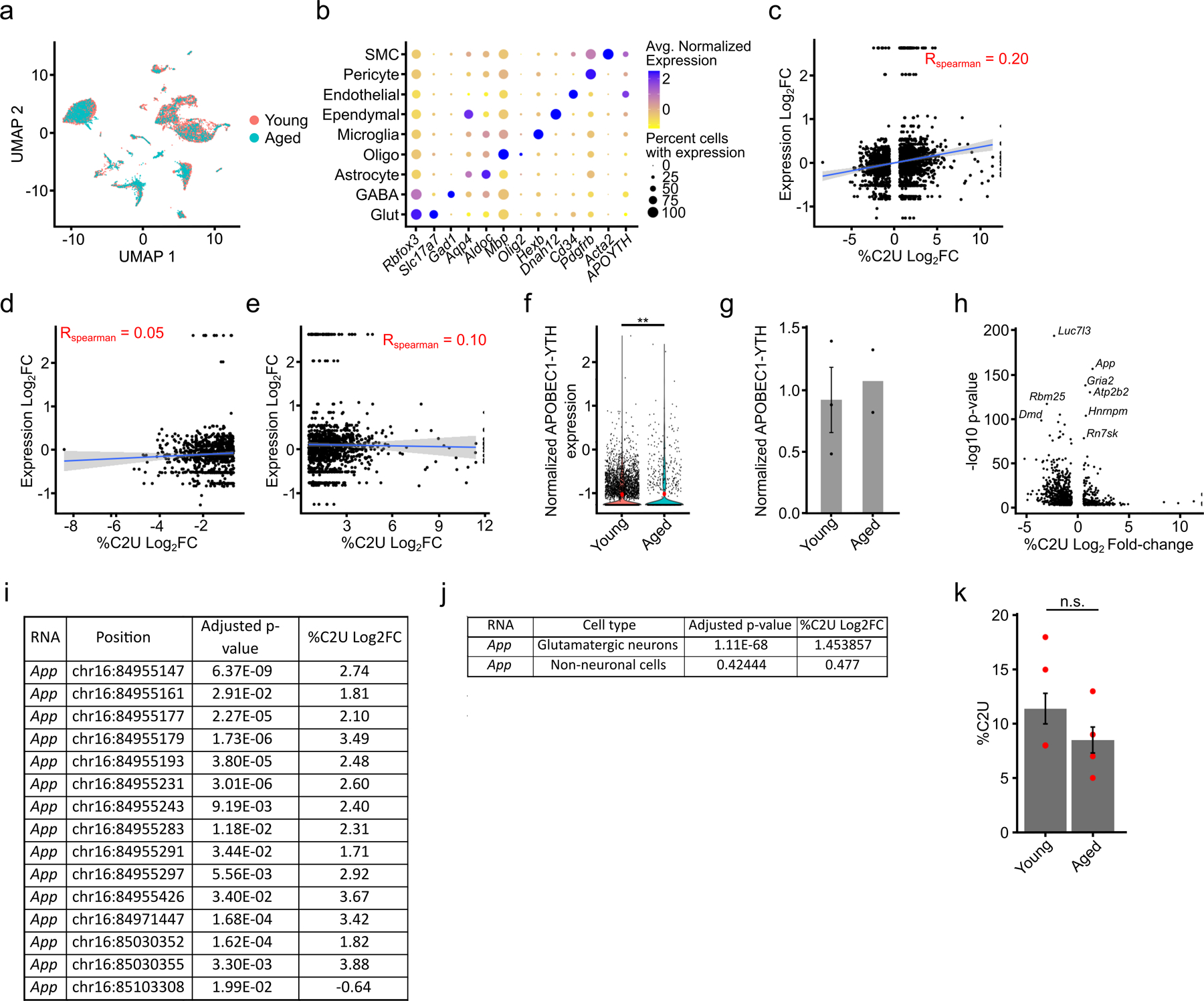
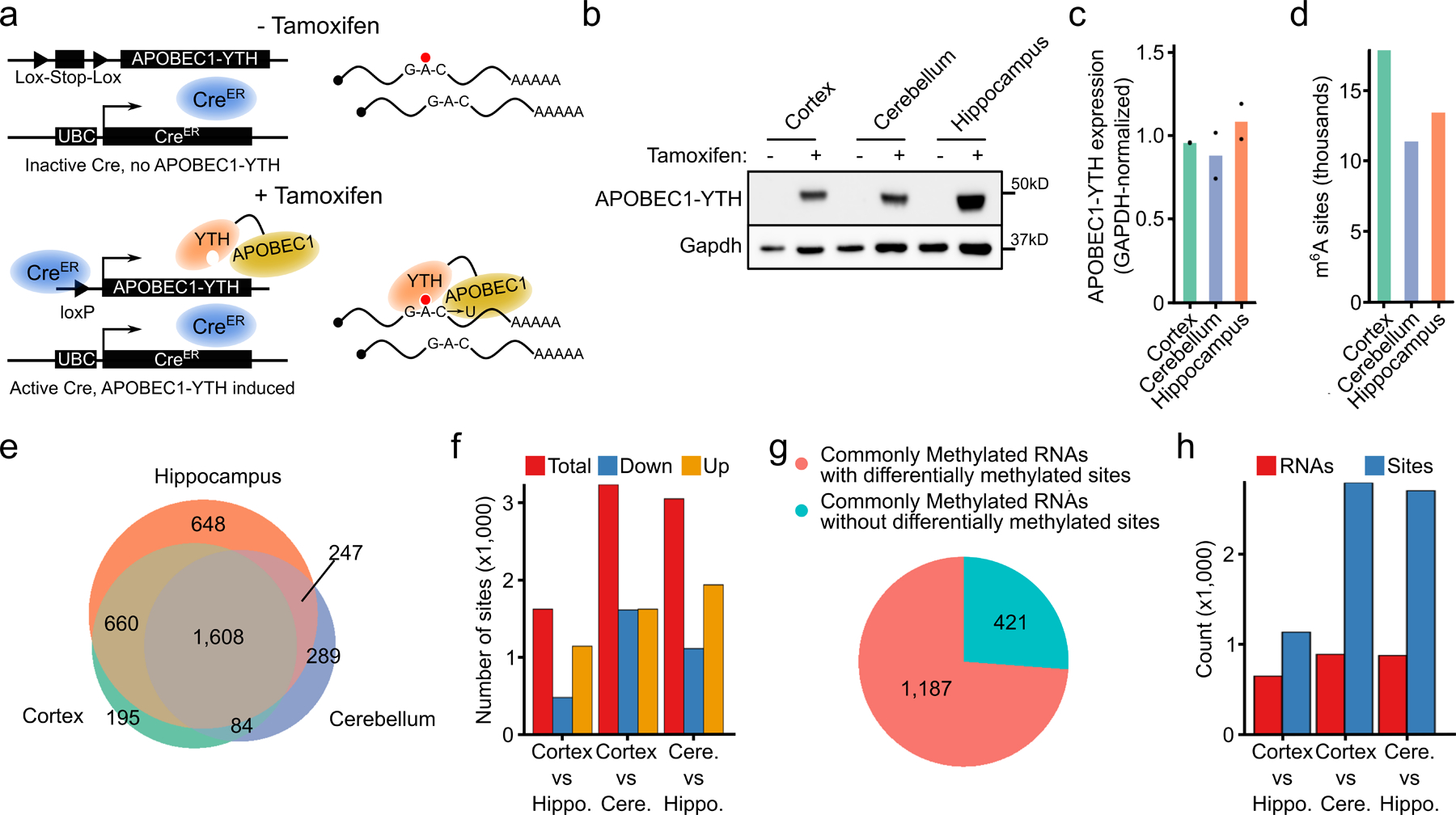
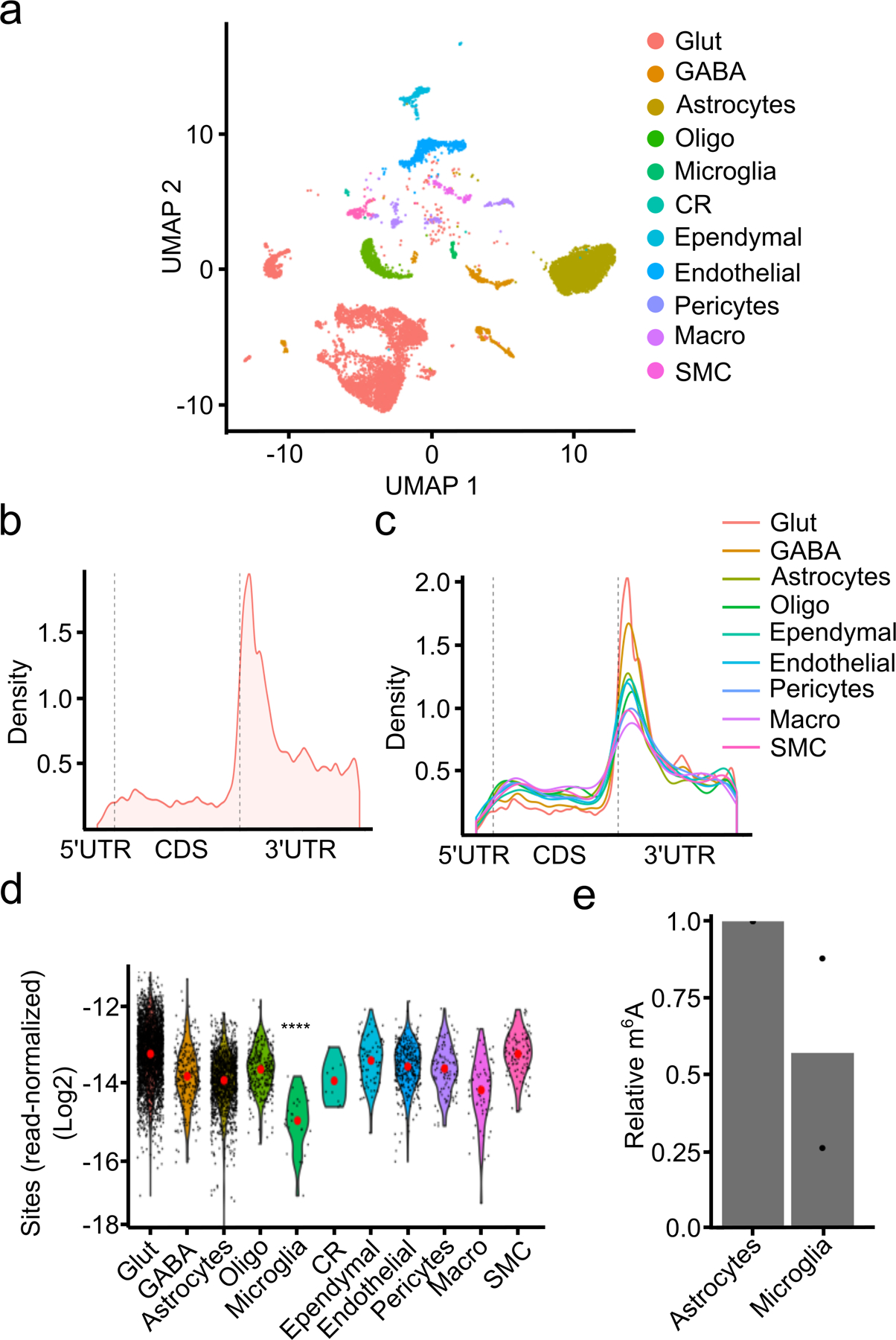
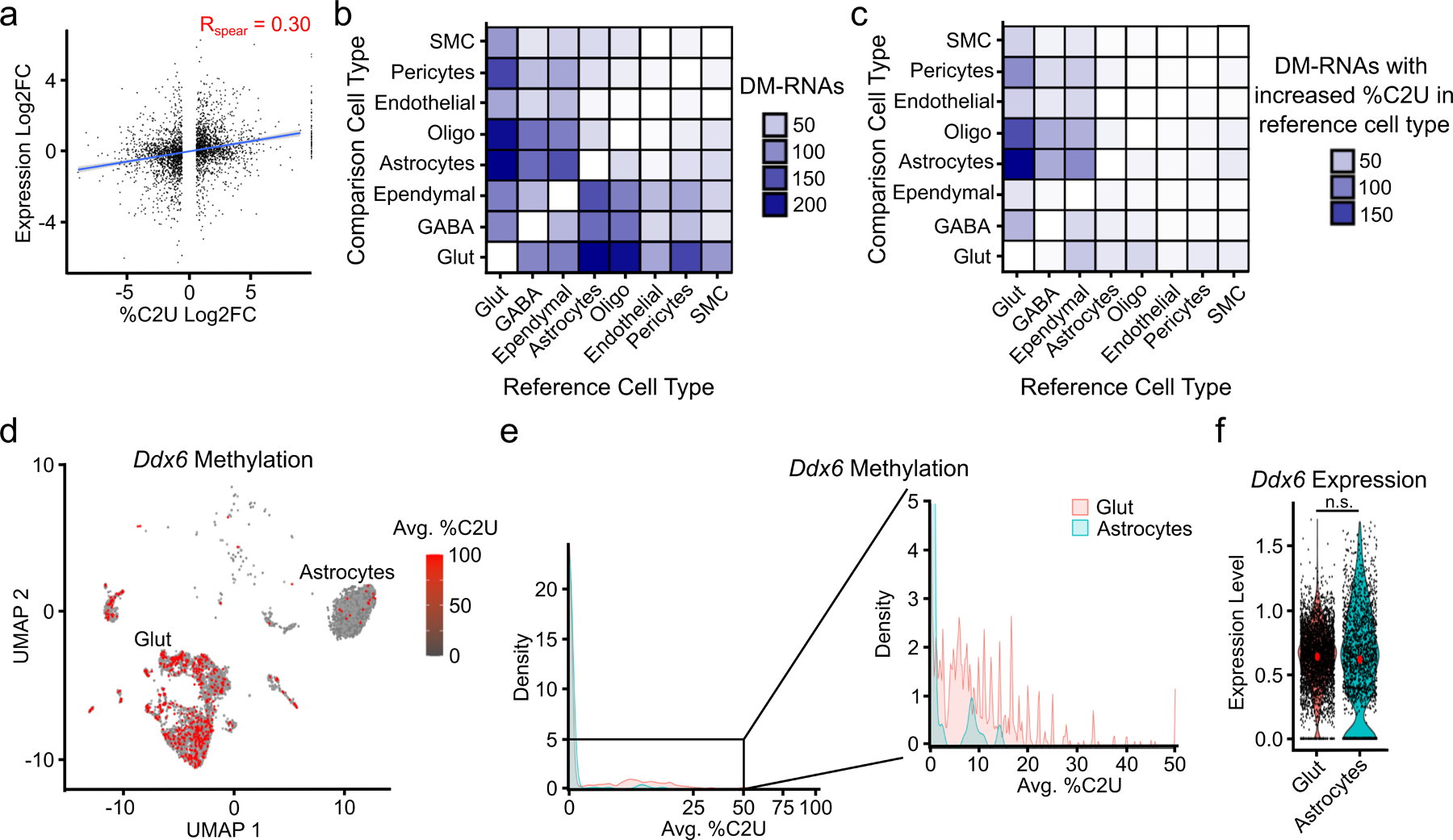
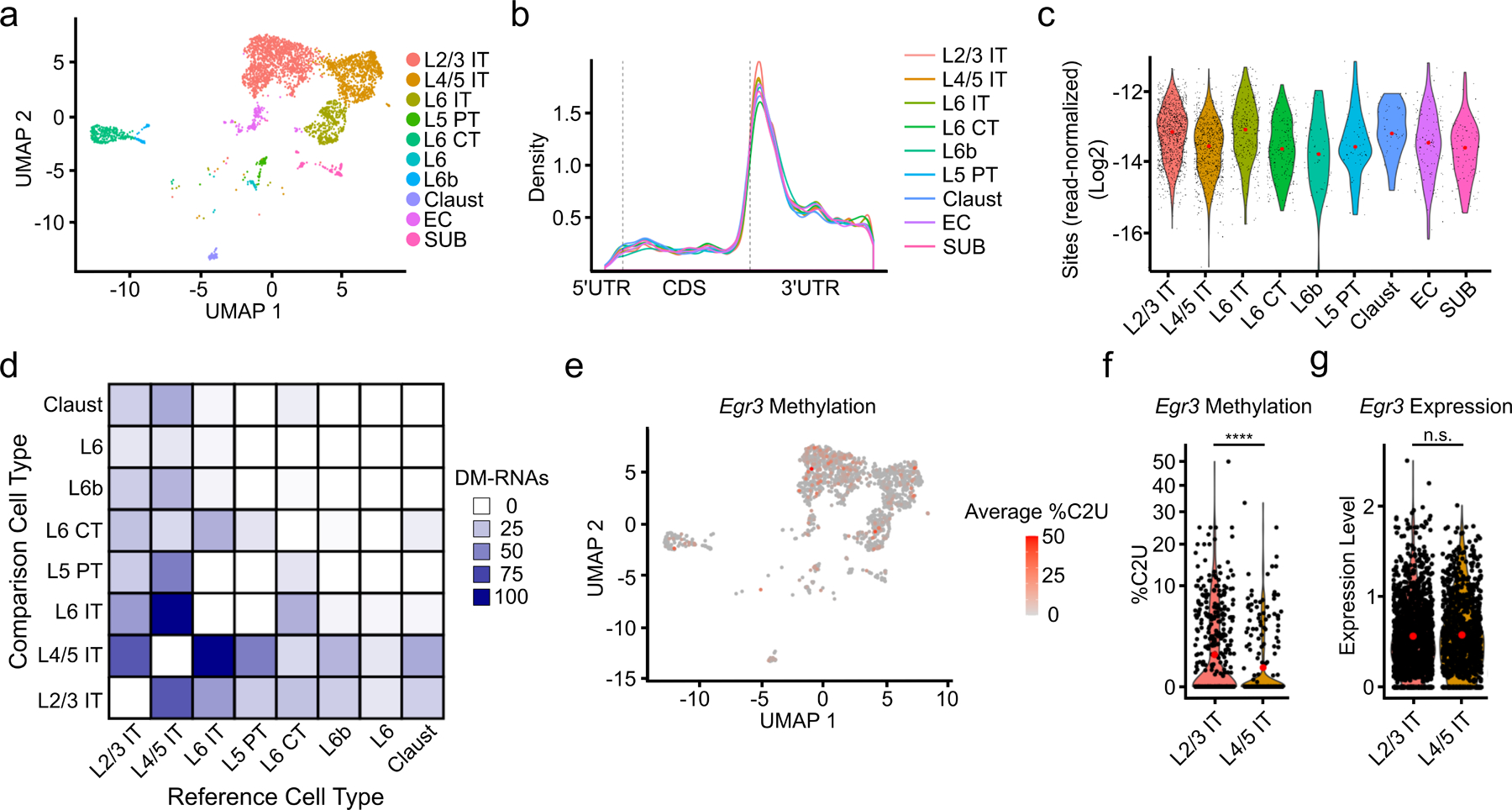
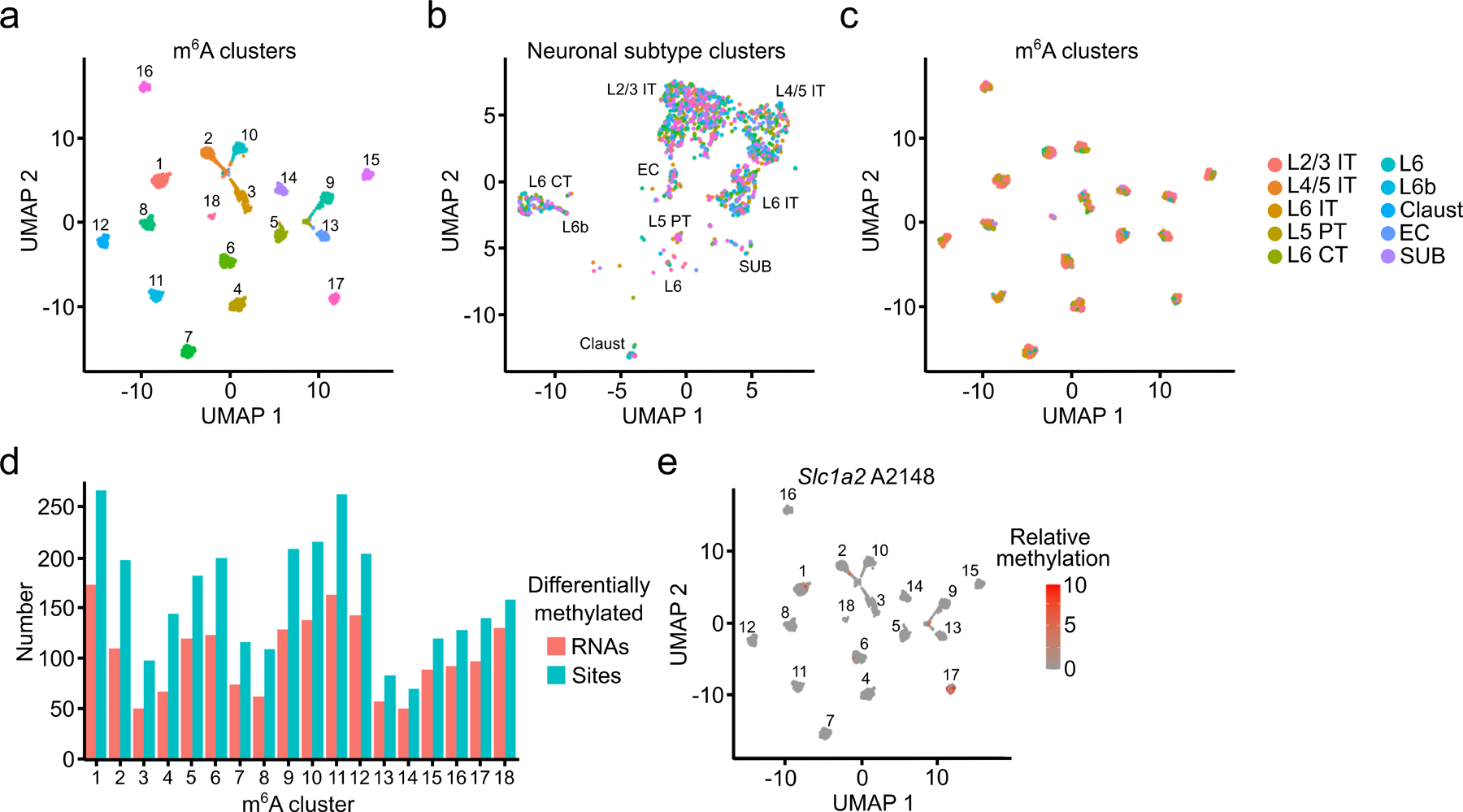
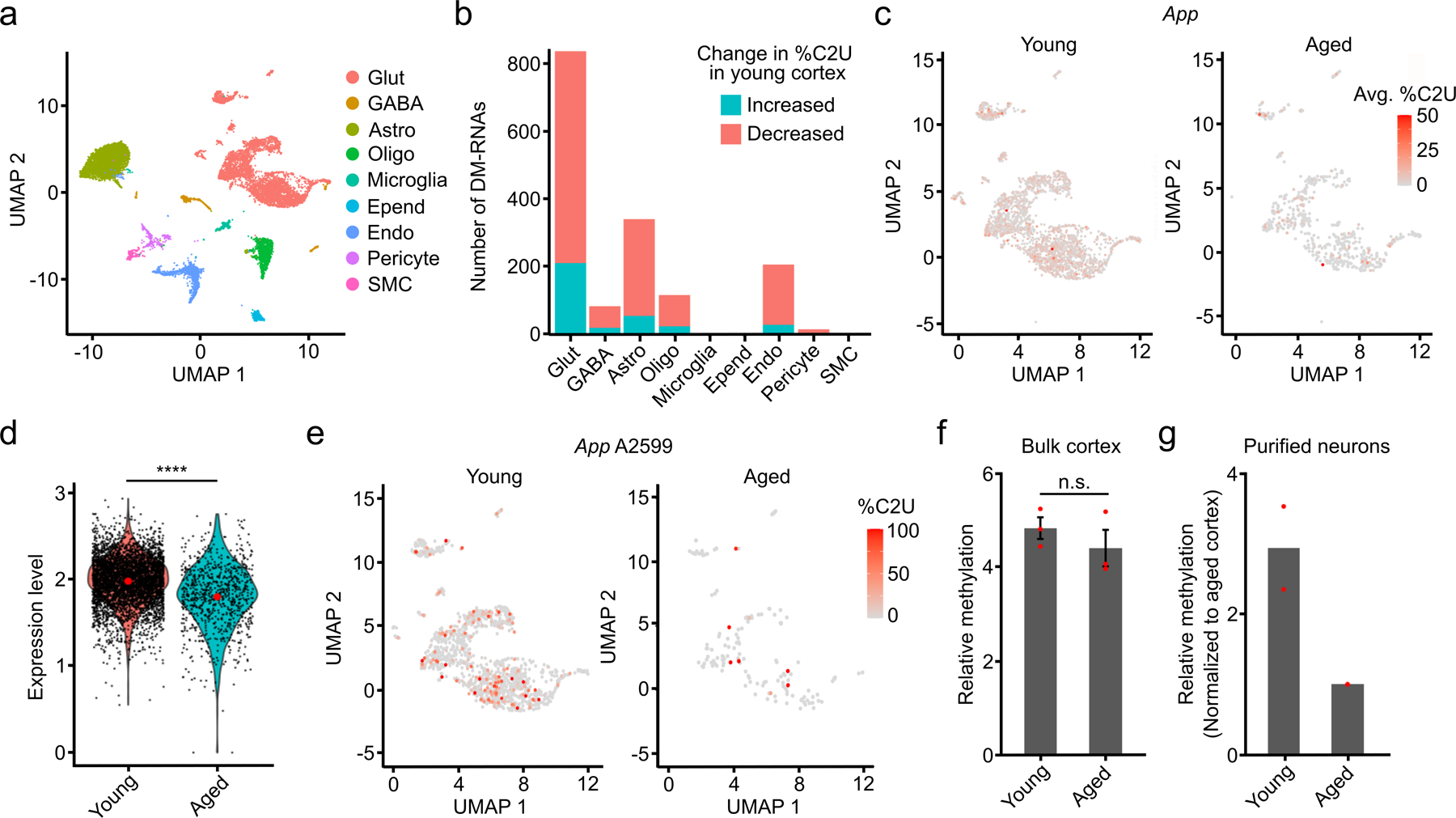
N6-methyladenosine (m6A) is an abundant mRNA modification in the brain that has important roles in neurodevelopment and brain function. However, because of technical limitations, global profiling of m6A sites within the individual cell types that make up the brain has not been possible. Here, we develop a mouse model that enables transcriptome-wide m6A detection in any tissue of interest at single-cell resolution. We use these mice to map m6A across different brain regions and within single cells of the mouse cortex and discover a high degree of shared methylation across brain regions and cell types. However, we also identify a small number of differentially methylated mRNAs in neurons that encode important regulators of neuronal signaling, and we discover that microglia have lower levels of m6A than other cell types. Finally, we perform single-cell m6A mapping in aged mice and identify many transcripts with age-dependent changes in m6A.
© 2024. The Author(s), under exclusive licence to Springer Nature America, Inc.
Conflict of interest statement
Competing interests: K.D.M. has filed a patent application for the DART-seq technology through Duke University. All other authors declare no competing interests.
Figures
References
-
- Zaccara S, Ries RJ & Jaffrey SR Reading, writing and erasing mRNA methylation. Nat. Rev. Mol. Cell Biol 20, 608–624 (2019). - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
