

Single-cell multi-omics map of human fetal blood in Down syndrome
- PMID: 39322663
- PMCID: PMC11446839
- DOI: 10.1038/s41586-024-07946-4
Single-cell multi-omics map of human fetal blood in Down syndrome
Abstract
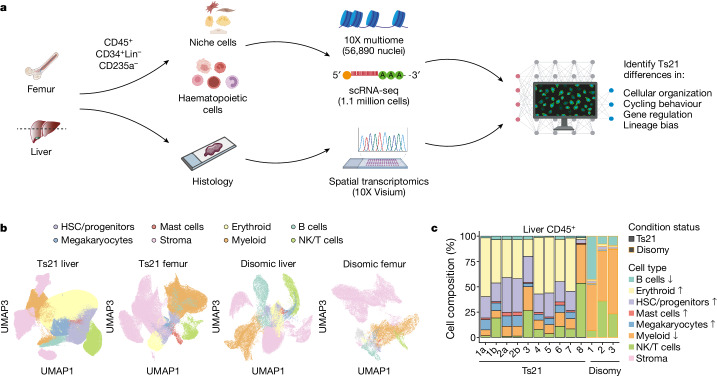
Down syndrome predisposes individuals to haematological abnormalities, such as increased number of erythrocytes and leukaemia in a process that is initiated before birth and is not entirely understood1-3. Here, to understand dysregulated haematopoiesis in Down syndrome, we integrated single-cell transcriptomics of over 1.1 million cells with chromatin accessibility and spatial transcriptomics datasets using human fetal liver and bone marrow samples from 3 fetuses with disomy and 15 fetuses with trisomy. We found that differences in gene expression in Down syndrome were dependent on both cell type and environment. Furthermore, we found multiple lines of evidence that haematopoietic stem cells (HSCs) in Down syndrome are 'primed' to differentiate. We subsequently established a Down syndrome-specific map linking non-coding elements to genes in disomic and trisomic HSCs using 10X multiome data. By integrating this map with genetic variants associated with blood cell counts, we discovered that trisomy restructured regulatory interactions to dysregulate enhancer activity and gene expression critical to erythroid lineage differentiation. Furthermore, as mutations in Down syndrome display a signature of oxidative stress4,5, we validated both increased mitochondrial mass and oxidative stress in Down syndrome, and observed that these mutations preferentially fell into regulatory regions of expressed genes in HSCs. Together, our single-cell, multi-omic resource provides a high-resolution molecular map of fetal haematopoiesis in Down syndrome and indicates significant regulatory restructuring giving rise to co-occurring haematological conditions.
© 2024. The Author(s).
Conflict of interest statement
A.R.M. consults for Third Rock Ventures, Inc. S.B.M. advises BioMarin, MyOme and Tenaya Therapeutics. All other authors declare no competing interests.
Figures











References
-
- Hasle, H., Clemmensen, I. H. & Mikkelsen, M. Risks of leukaemia and solid tumours in individuals with Down’s syndrome. Lancet355, 165–169 (2000). - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
