

The type 2 cytokine Fc-IL-4 revitalizes exhausted CD8+ T cells against cancer
- PMID: 39322665
- PMCID: PMC11485240
- DOI: 10.1038/s41586-024-07962-4
The type 2 cytokine Fc-IL-4 revitalizes exhausted CD8+ T cells against cancer
Abstract
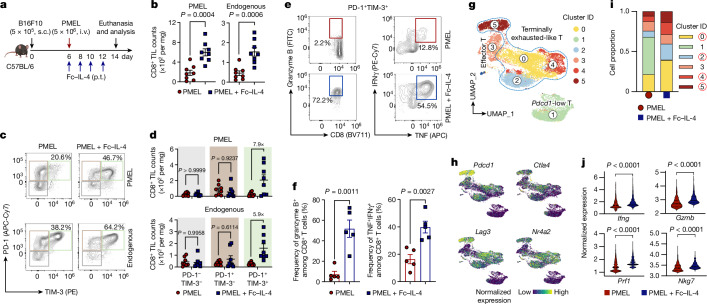
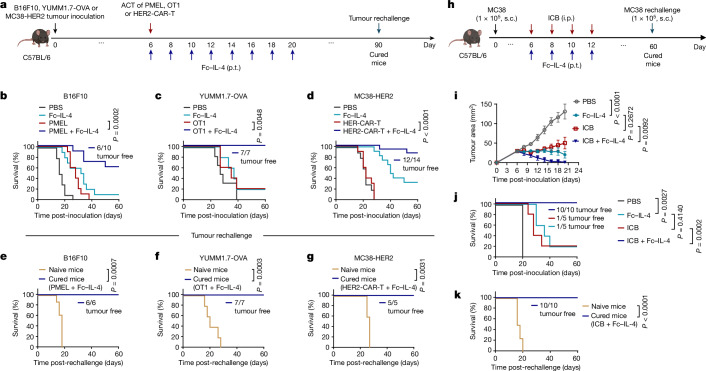
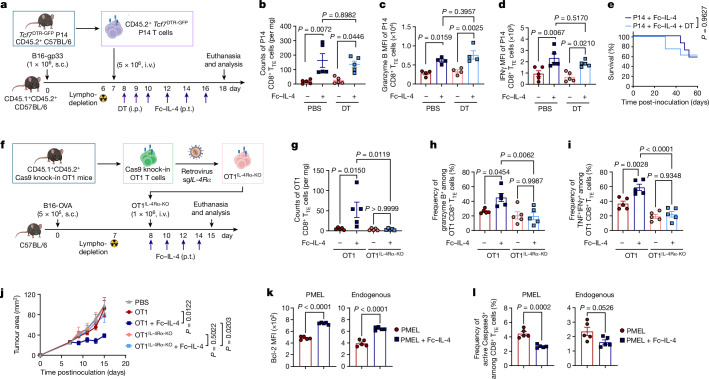
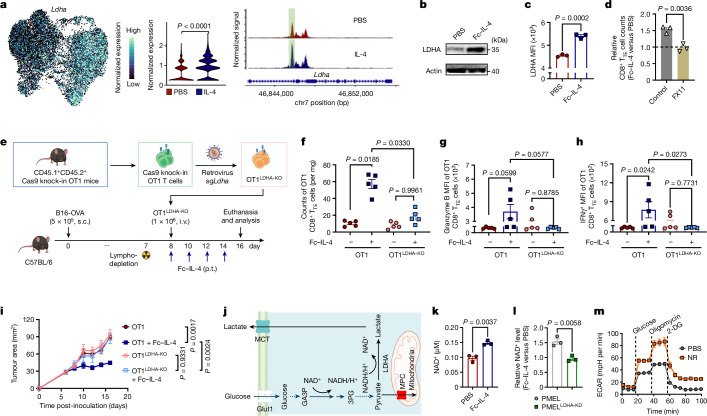
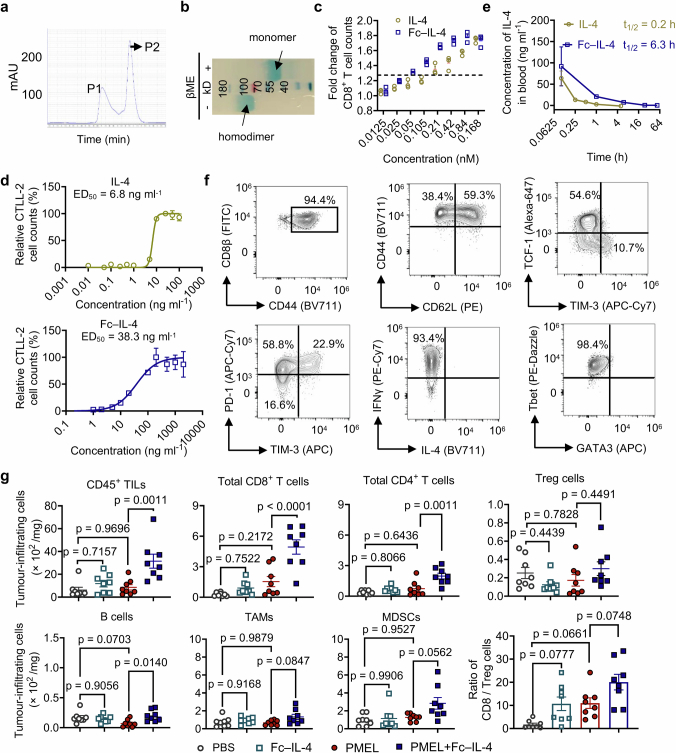
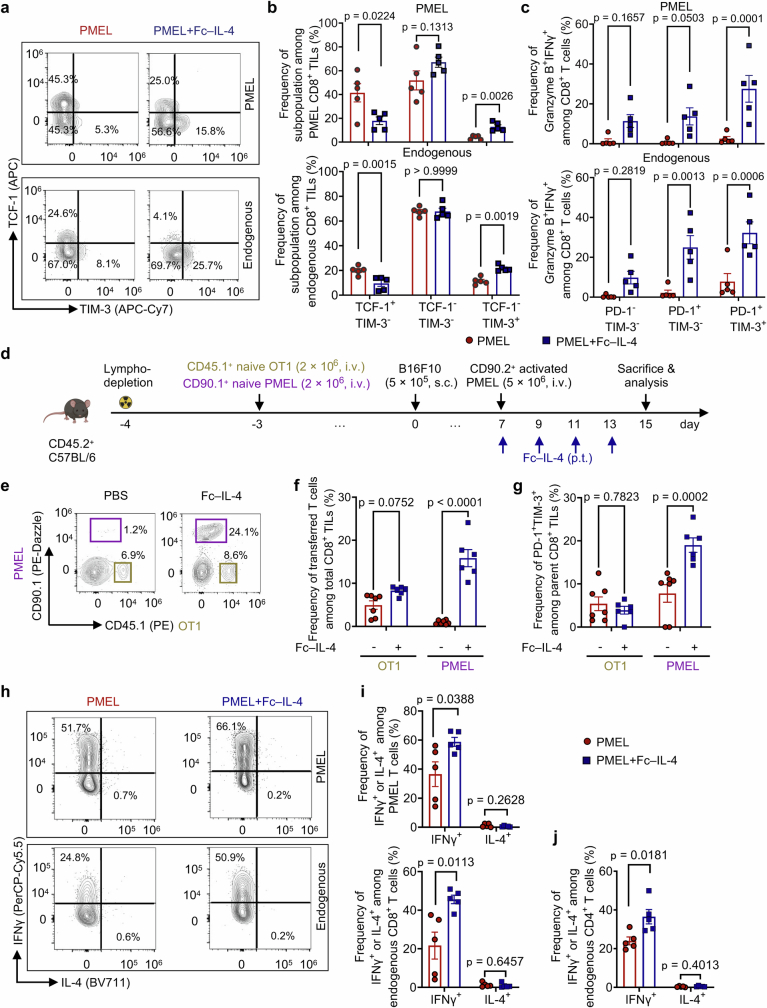
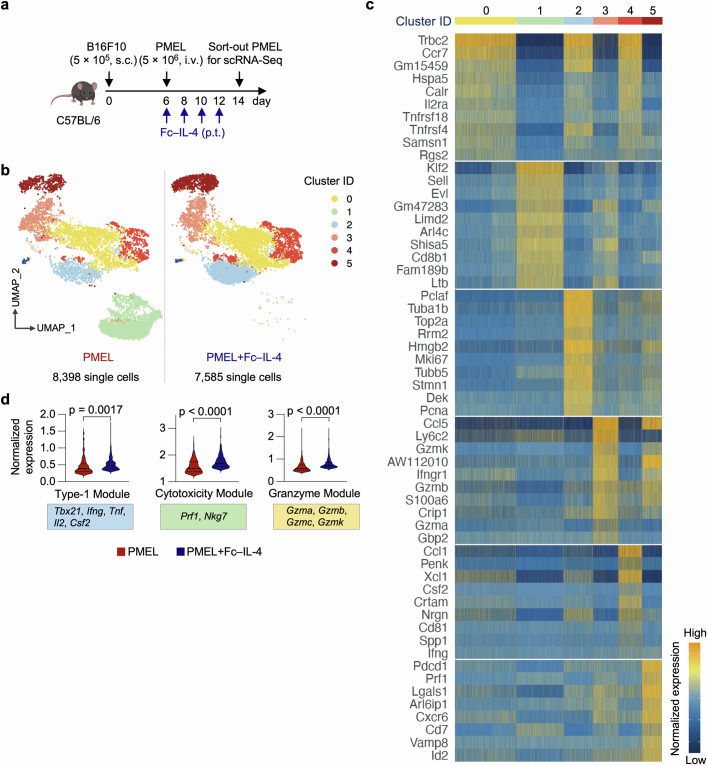
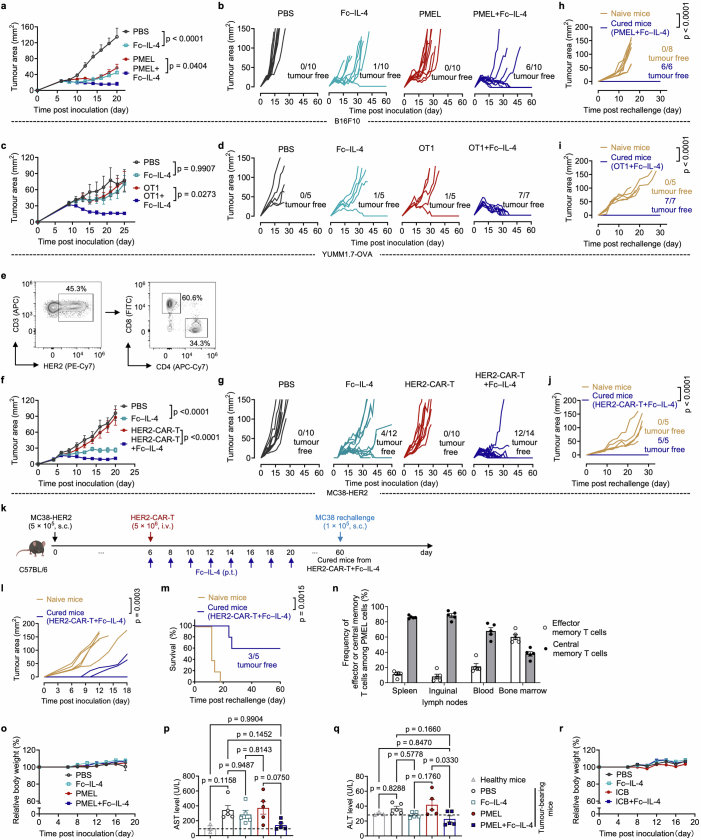
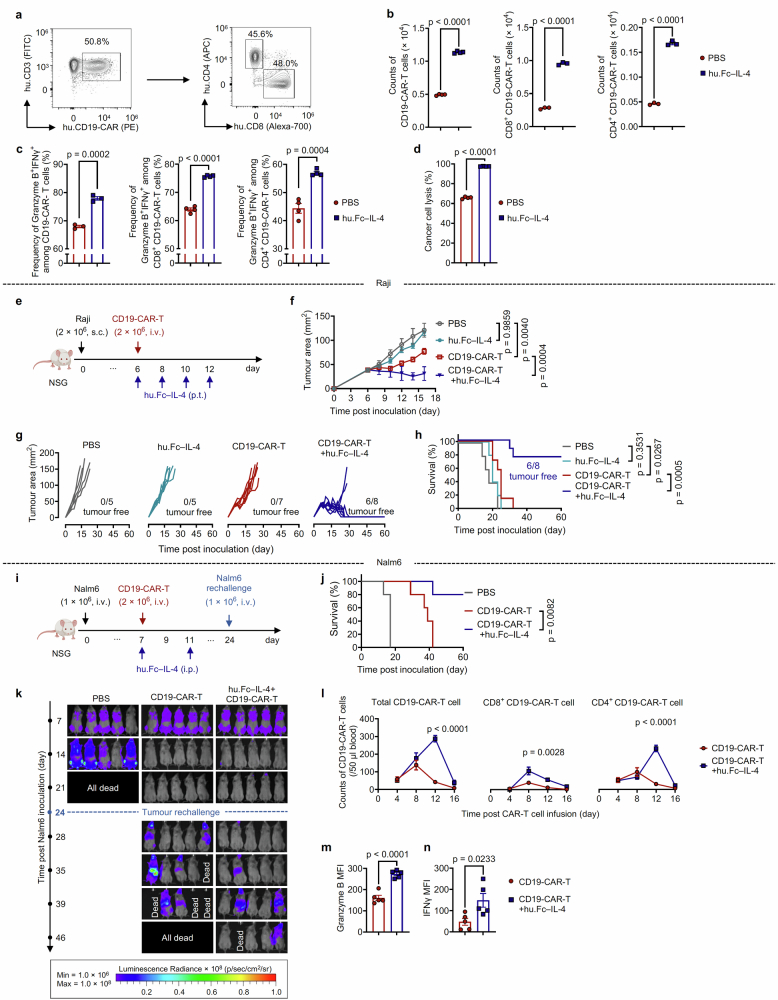
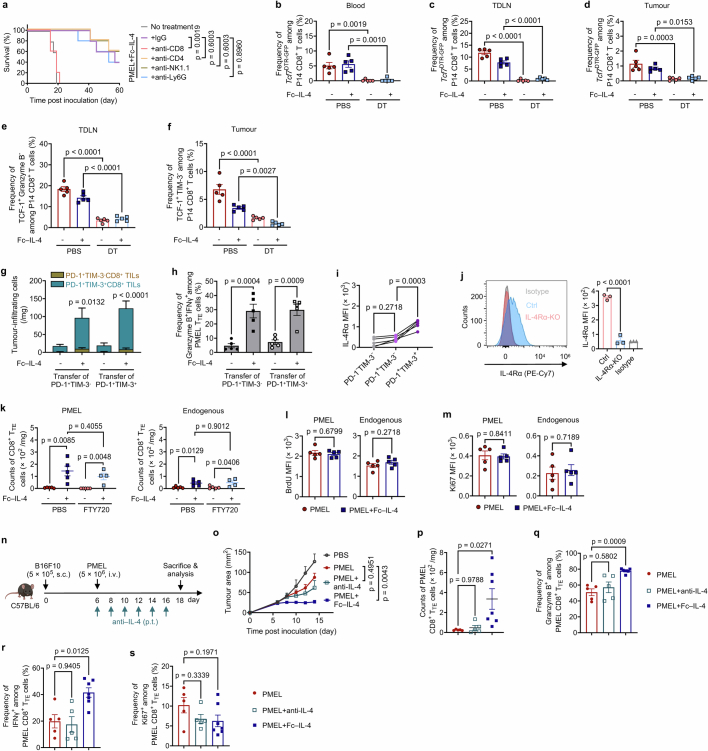
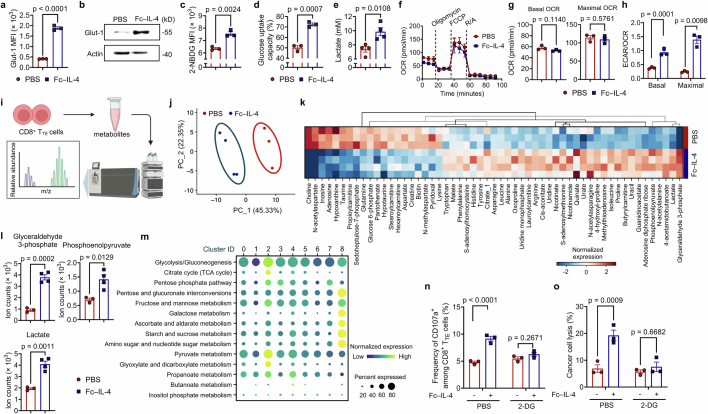
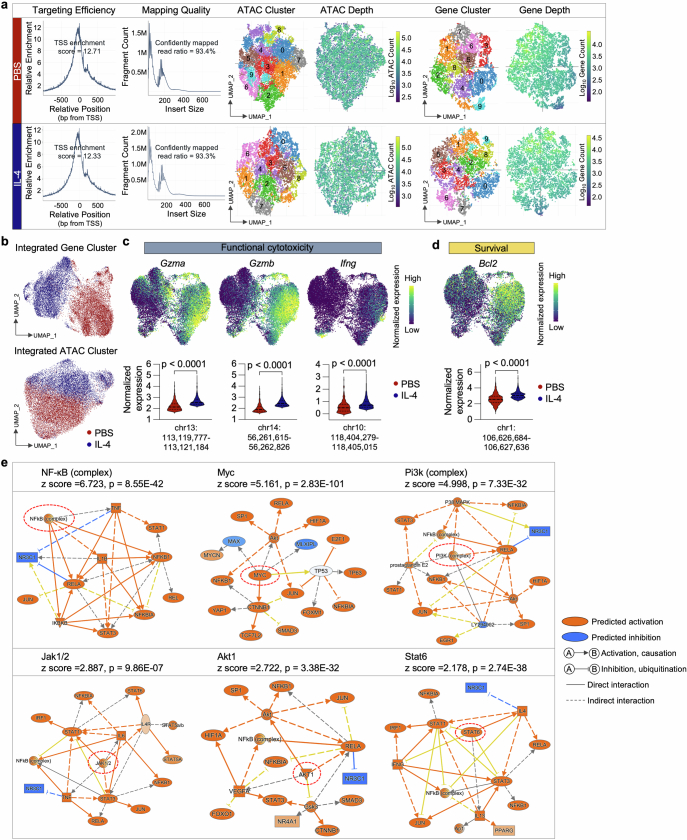
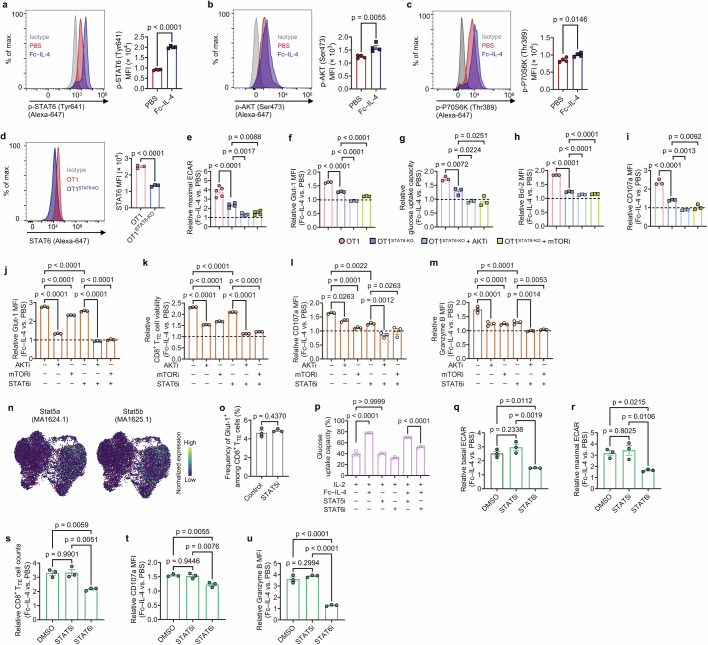
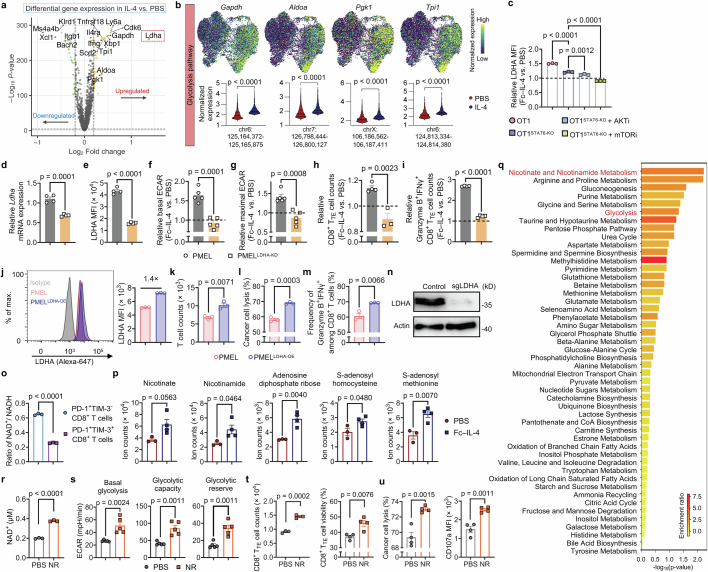
Current cancer immunotherapy predominately focuses on eliciting type 1 immune responses fighting cancer; however, long-term complete remission remains uncommon1,2. A pivotal question arises as to whether type 2 immunity can be orchestrated alongside type 1-centric immunotherapy to achieve enduring response against cancer3,4. Here we show that an interleukin-4 fusion protein (Fc-IL-4), a typical type 2 cytokine, directly acts on CD8+ T cells and enriches functional terminally exhausted CD8+ T (CD8+ TTE) cells in the tumour. Consequently, Fc-IL-4 enhances antitumour efficacy of type 1 immunity-centric adoptive T cell transfer or immune checkpoint blockade therapies and induces durable remission across several syngeneic and xenograft tumour models. Mechanistically, we discovered that Fc-IL-4 signals through both signal transducer and activator of transcription 6 (STAT6) and mammalian target of rapamycin (mTOR) pathways, augmenting the glycolytic metabolism and the nicotinamide adenine dinucleotide (NAD) concentration of CD8+ TTE cells in a lactate dehydrogenase A-dependent manner. The metabolic modulation mediated by Fc-IL-4 is indispensable for reinvigorating intratumoural CD8+ TTE cells. These findings underscore Fc-IL-4 as a potent type 2 cytokine-based immunotherapy that synergizes effectively with type 1 immunity to elicit long-lasting responses against cancer. Our study not only sheds light on the synergy between these two types of immune responses, but also unveils an innovative strategy for advancing next-generation cancer immunotherapy by integrating type 2 immune factors.
© 2024. The Author(s).
Conflict of interest statement
Y.G., L.T. and B.F. are inventors of the patents relevant to the findings reported here. L.T. and Y.G. are cofounders, share-holders and advisors for Leman Biotech. The interests of L.T. were reviewed and managed by EPFL. R.F. is scientific founder and adviser for IsoPlexis, Singleron Biotechnologies and AtlasXomics. The interests of R.F. were reviewed and managed by Yale University Provost’s Office in accordance with the University’s conflict of interest policies. J.J.M. hold patents related to CAR-T cell manufacturing and biomarker discovery. The remaining authors declare no competing interests.
Figures

References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
