

The genetic architecture of protein stability
- PMID: 39322666
- PMCID: PMC11499273
- DOI: 10.1038/s41586-024-07966-0
The genetic architecture of protein stability
Abstract
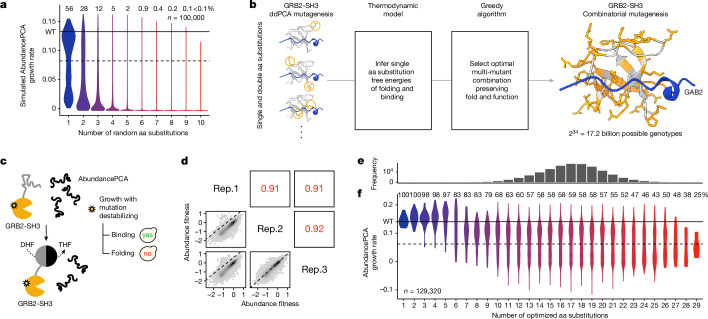
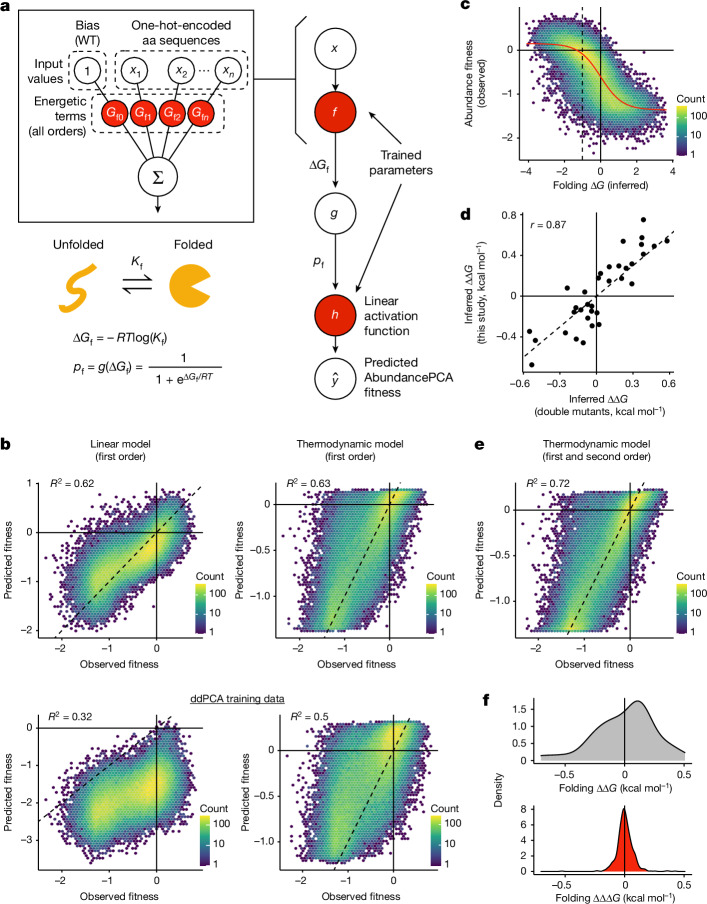
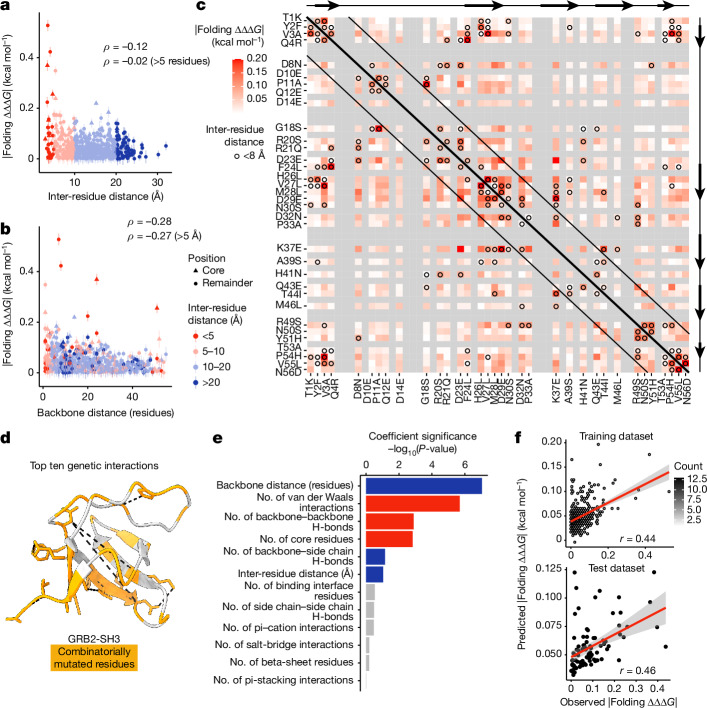
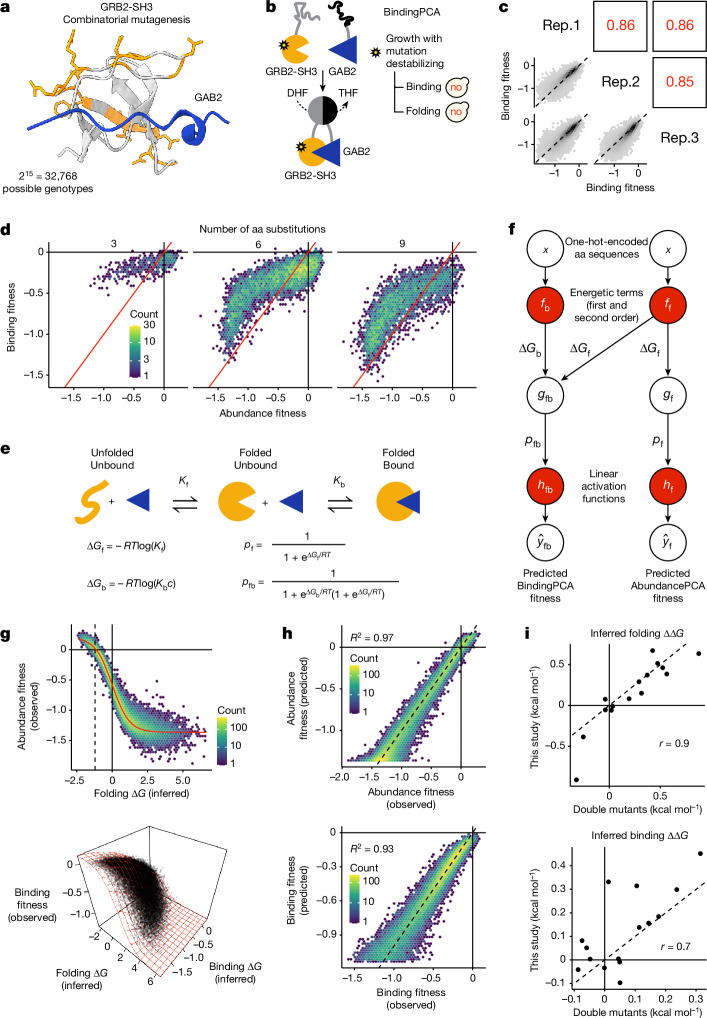
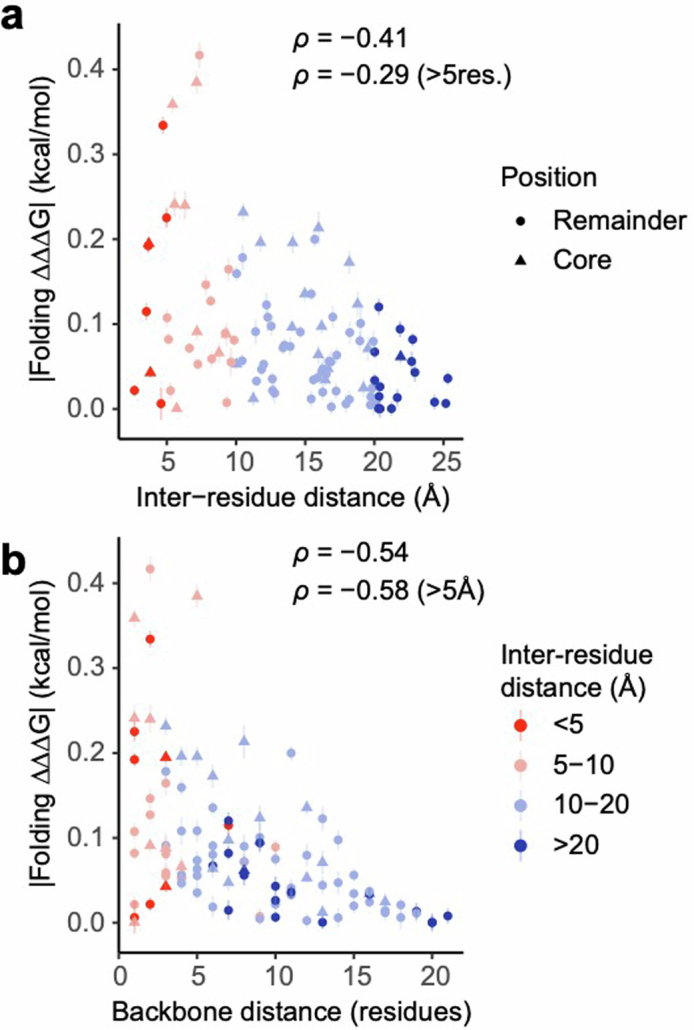
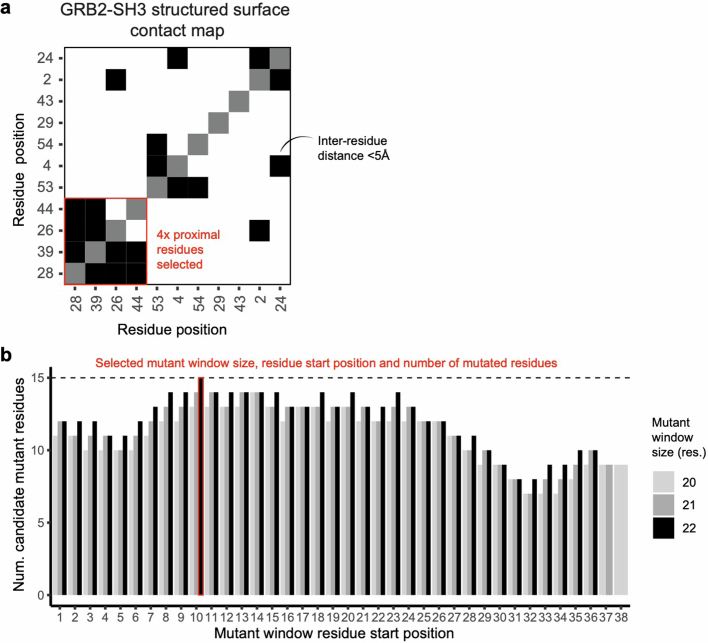
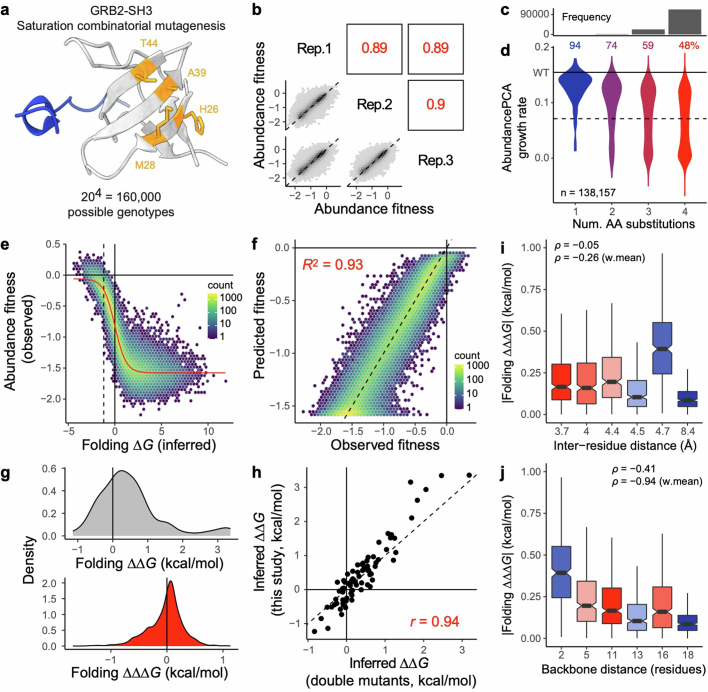
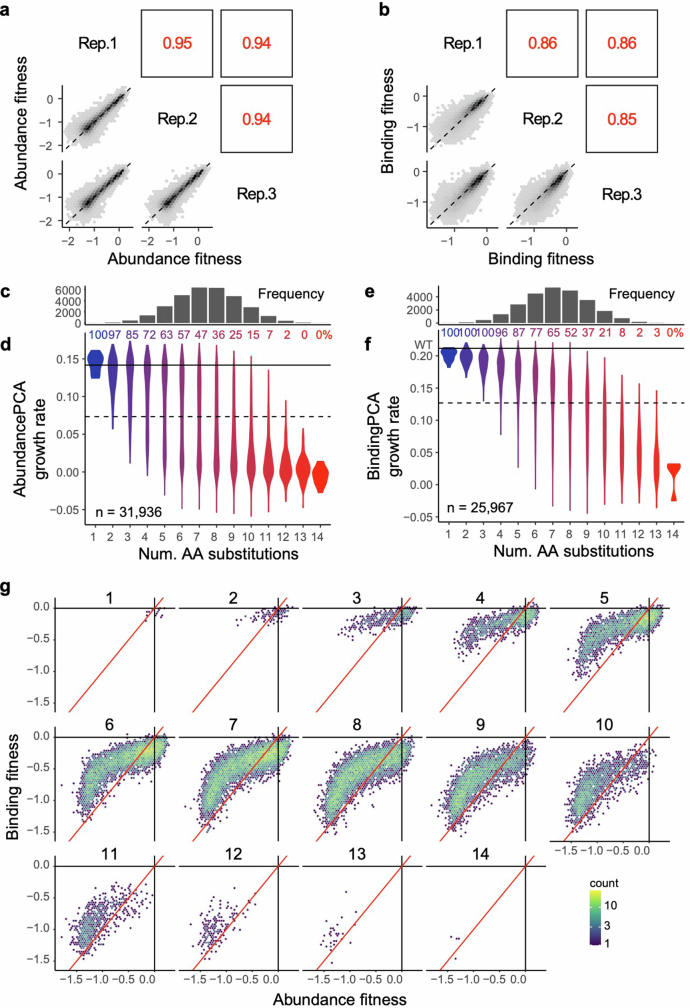
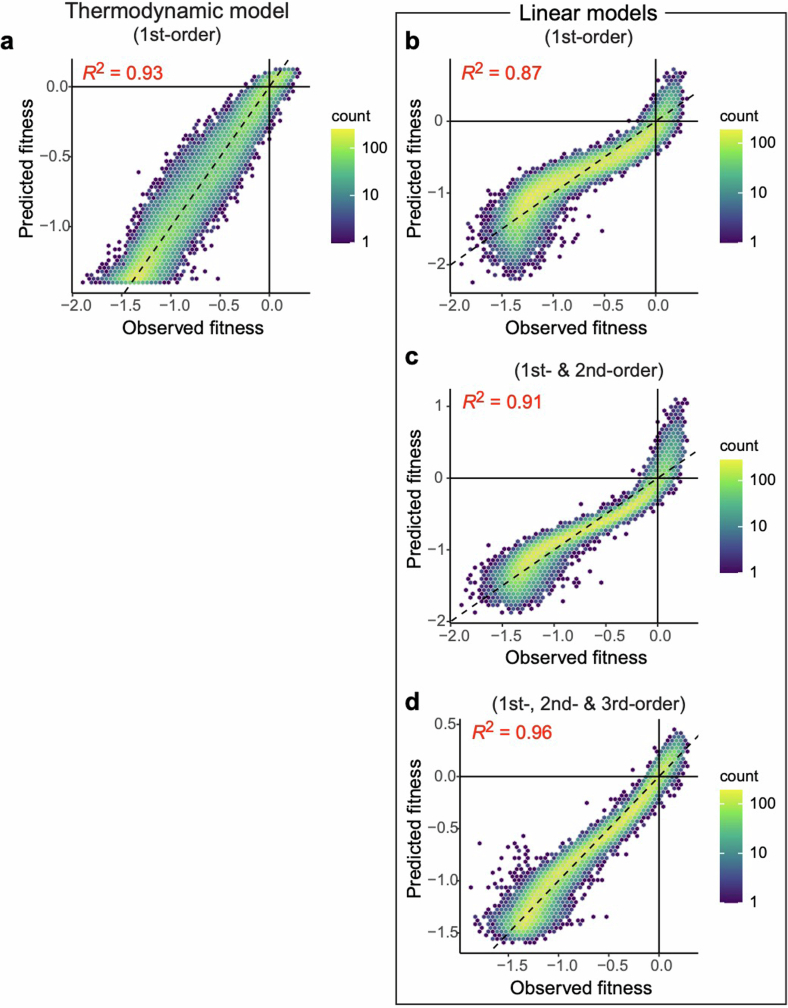
There are more ways to synthesize a 100-amino acid (aa) protein (20100) than there are atoms in the universe. Only a very small fraction of such a vast sequence space can ever be experimentally or computationally surveyed. Deep neural networks are increasingly being used to navigate high-dimensional sequence spaces1. However, these models are extremely complicated. Here, by experimentally sampling from sequence spaces larger than 1010, we show that the genetic architecture of at least some proteins is remarkably simple, allowing accurate genetic prediction in high-dimensional sequence spaces with fully interpretable energy models. These models capture the nonlinear relationships between free energies and phenotypes but otherwise consist of additive free energy changes with a small contribution from pairwise energetic couplings. These energetic couplings are sparse and associated with structural contacts and backbone proximity. Our results indicate that protein genetics is actually both rather simple and intelligible.
© 2024. The Author(s).
Conflict of interest statement
A.J.F. and B.L. are founders, employees and shareholders of ALLOX. J.M.S. is a founder, employee and shareholder of factorize.bio.
Figures
References
-
- Notin, P., Rollins, N., Gal, Y., Sander, C. & Marks, D. Machine learning for functional protein design. Nat. Biotechnol.42, 216–228 (2024). - PubMed
-
- Kinney, J. B. & McCandlish, D. M. Massively parallel assays and quantitative sequence–function relationships. Annu. Rev. Genom. Hum. Genet.20, 99–127 (2019). - PubMed
-
- Nedrud, D., Coyote-Maestas, W. & Schmidt, D. A large-scale survey of pairwise epistasis reveals a mechanism for evolutionary expansion and specialization of PDZ domains. Proteins89, 899–914 (2021). - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
