

Multistate and functional protein design using RoseTTAFold sequence space diffusion
- PMID: 39322764
- PMCID: PMC12339374
- DOI: 10.1038/s41587-024-02395-w
Multistate and functional protein design using RoseTTAFold sequence space diffusion
Erratum in
-
Publisher Correction: Multistate and functional protein design using RoseTTAFold sequence space diffusion.Nat Biotechnol. 2025 Aug;43(8):1384. doi: 10.1038/s41587-024-02456-0. Nat Biotechnol. 2025. PMID: 39375454 Free PMC article. No abstract available.
Abstract
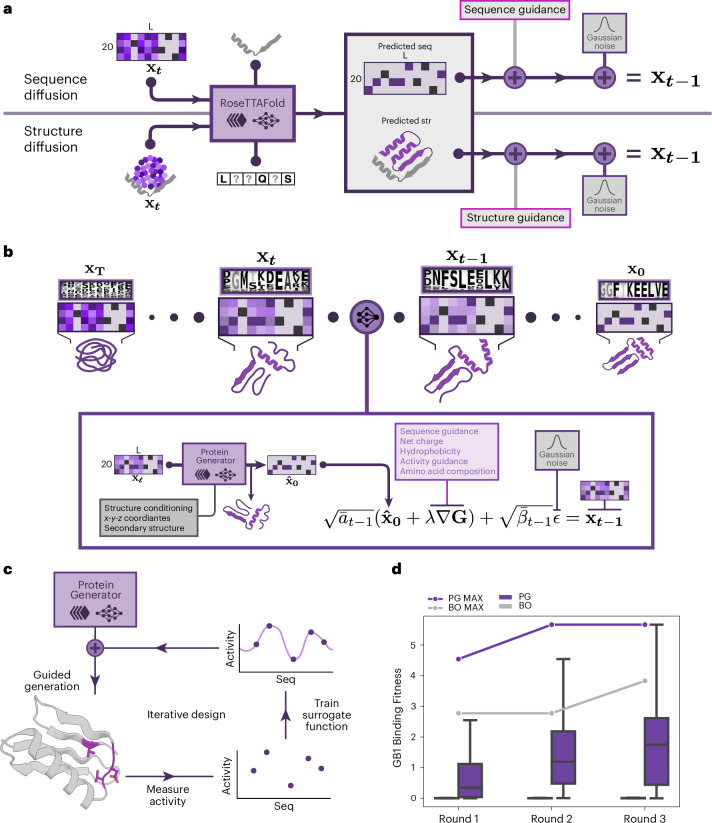
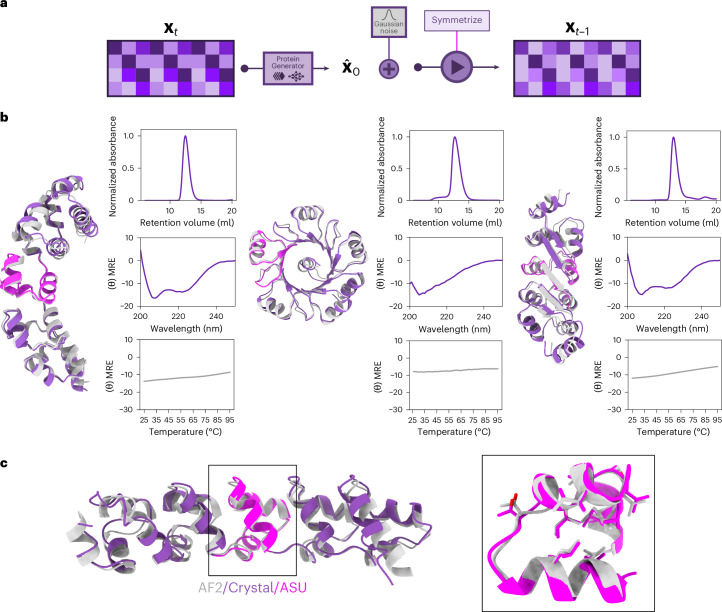
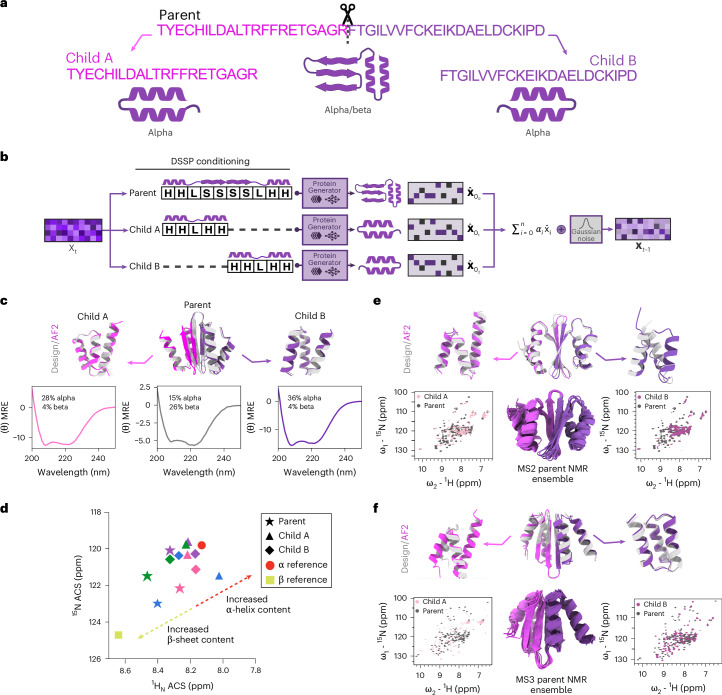
Protein denoising diffusion probabilistic models are used for the de novo generation of protein backbones but are limited in their ability to guide generation of proteins with sequence-specific attributes and functional properties. To overcome this limitation, we developed ProteinGenerator (PG), a sequence space diffusion model based on RoseTTAFold that simultaneously generates protein sequences and structures. Beginning from a noised sequence representation, PG generates sequence and structure pairs by iterative denoising, guided by desired sequence and structural protein attributes. We designed thermostable proteins with varying amino acid compositions and internal sequence repeats and cage bioactive peptides, such as melittin. By averaging sequence logits between diffusion trajectories with distinct structural constraints, we designed multistate parent-child protein triples in which the same sequence folds to different supersecondary structures when intact in the parent versus split into two child domains. PG design trajectories can be guided by experimental sequence-activity data, providing a general approach for integrated computational and experimental optimization of protein function.
© 2024. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures


Similar articles
-
Prescription of Controlled Substances: Benefits and Risks.2025 Jul 6. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. 2025 Jul 6. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. PMID: 30726003 Free Books & Documents.
-
Electrophoresis.2025 Jul 14. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. 2025 Jul 14. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. PMID: 36251838 Free Books & Documents.
-
Short-Term Memory Impairment.2024 Jun 8. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. 2024 Jun 8. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. PMID: 31424720 Free Books & Documents.
-
The effect of sample site and collection procedure on identification of SARS-CoV-2 infection.Cochrane Database Syst Rev. 2024 Dec 16;12(12):CD014780. doi: 10.1002/14651858.CD014780. Cochrane Database Syst Rev. 2024. PMID: 39679851 Free PMC article.
-
A rapid and systematic review of the clinical effectiveness and cost-effectiveness of paclitaxel, docetaxel, gemcitabine and vinorelbine in non-small-cell lung cancer.Health Technol Assess. 2001;5(32):1-195. doi: 10.3310/hta5320. Health Technol Assess. 2001. PMID: 12065068
Cited by
-
Engineering Dehalogenase Enzymes Using Variational Autoencoder-Generated Latent Spaces and Microfluidics.JACS Au. 2025 Feb 13;5(2):838-850. doi: 10.1021/jacsau.4c01101. eCollection 2025 Feb 24. JACS Au. 2025. PMID: 40017771 Free PMC article.
-
Supervised fine-tuning of pre-trained antibody language models improves antigen specificity prediction.PLoS Comput Biol. 2025 Mar 31;21(3):e1012153. doi: 10.1371/journal.pcbi.1012153. eCollection 2025 Mar. PLoS Comput Biol. 2025. PMID: 40163503 Free PMC article.
-
Rational engineering of allosteric protein switches by in silico prediction of domain insertion sites.Nat Methods. 2025 Aug;22(8):1698-1706. doi: 10.1038/s41592-025-02741-z. Epub 2025 Aug 4. Nat Methods. 2025. PMID: 40759748 Free PMC article.
-
Discovery, design, and engineering of enzymes based on molecular retrobiosynthesis.mLife. 2025 Mar 28;4(2):107-125. doi: 10.1002/mlf2.70009. eCollection 2025 Apr. mLife. 2025. PMID: 40313979 Free PMC article. Review.
-
Ligand-Induced Biased Activation of GPCRs: Recent Advances and New Directions from In Silico Approaches.Molecules. 2025 Feb 25;30(5):1047. doi: 10.3390/molecules30051047. Molecules. 2025. PMID: 40076272 Free PMC article. Review.
References
-
- Winnifrith, A., Outeiral, C. & Hie, B. Generative artificial intelligence for de novo protein design. Preprint at arXiv10.48550/arXiv.2310.09685 (2023). - PubMed
-
- Notin, P., Rollins, N., Gal, Y., Sander, C. & Marks, D. Machine learning for functional protein design. Nat. Biotechnol.42, 216–228 (2024). - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
