

A realistic benchmark for differential abundance testing and confounder adjustment in human microbiome studies
- PMID: 39322959
- PMCID: PMC11423519
- DOI: 10.1186/s13059-024-03390-9
A realistic benchmark for differential abundance testing and confounder adjustment in human microbiome studies
Abstract
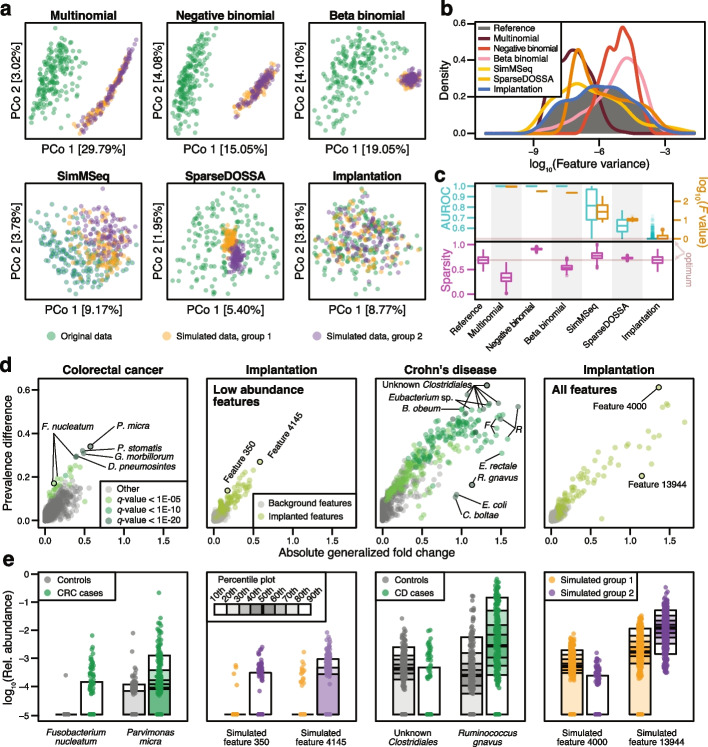
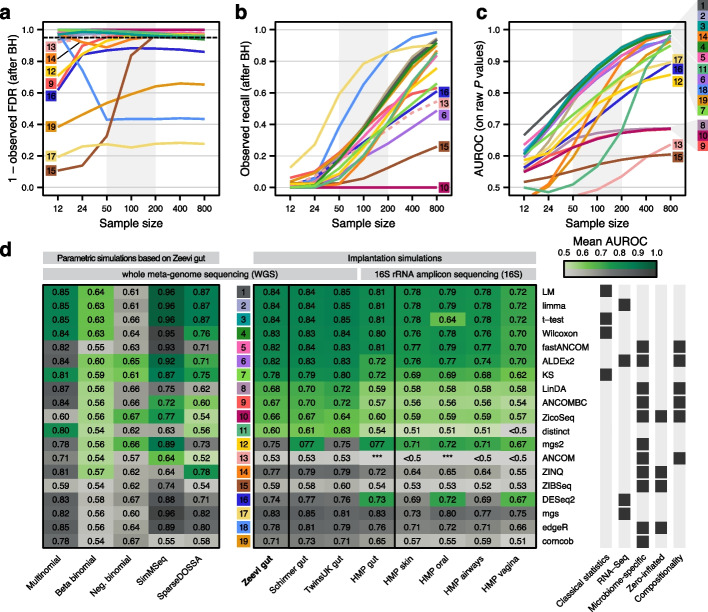
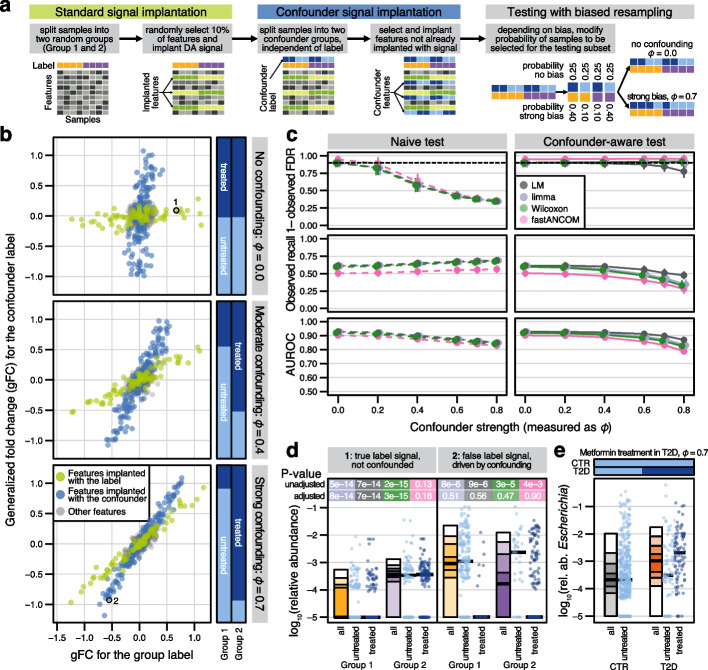
Background: In microbiome disease association studies, it is a fundamental task to test which microbes differ in their abundance between groups. Yet, consensus on suitable or optimal statistical methods for differential abundance testing is lacking, and it remains unexplored how these cope with confounding. Previous differential abundance benchmarks relying on simulated datasets did not quantitatively evaluate the similarity to real data, which undermines their recommendations.
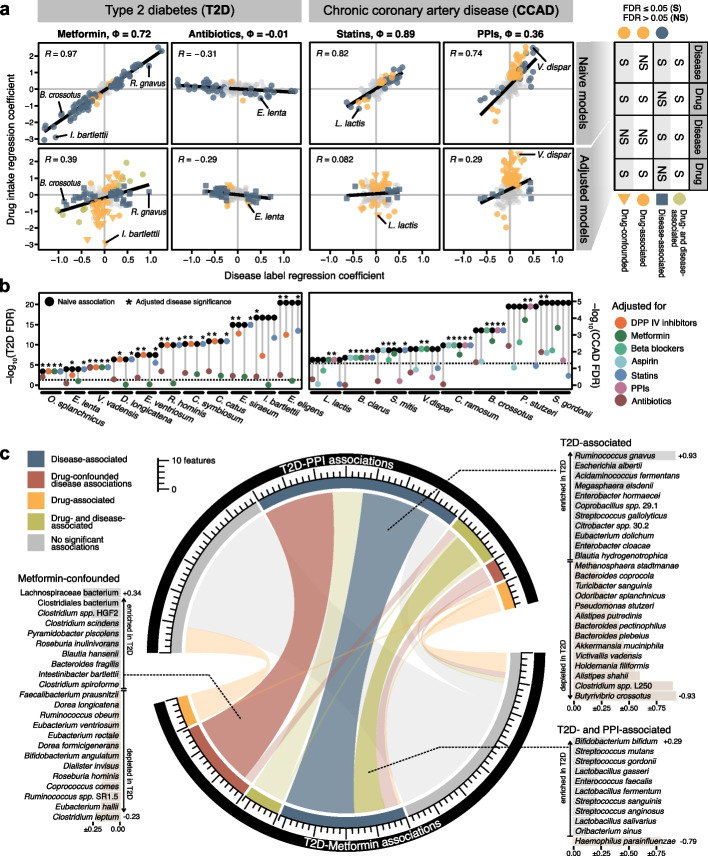
Results: Our simulation framework implants calibrated signals into real taxonomic profiles, including signals mimicking confounders. Using several whole meta-genome and 16S rRNA gene amplicon datasets, we validate that our simulated data resembles real data from disease association studies much more than in previous benchmarks. With extensively parametrized simulations, we benchmark the performance of nineteen differential abundance methods and further evaluate the best ones on confounded simulations. Only classic statistical methods (linear models, the Wilcoxon test, t-test), limma, and fastANCOM properly control false discoveries at relatively high sensitivity. When additionally considering confounders, these issues are exacerbated, but we find that adjusted differential abundance testing can effectively mitigate them. In a large cardiometabolic disease dataset, we showcase that failure to account for covariates such as medication causes spurious association in real-world applications.
Conclusions: Tight error control is critical for microbiome association studies. The unsatisfactory performance of many differential abundance methods and the persistent danger of unchecked confounding suggest these contribute to a lack of reproducibility among such studies. We have open-sourced our simulation and benchmarking software to foster a much-needed consolidation of statistical methodology for microbiome research.
Keywords: Benchmark; Confounding; Differential abundance; Metagenomics; Microbiome.
© 2024. The Author(s).
Conflict of interest statement
None of the authors declare any competing interests.
Figures
Similar articles
-
Computational Study Protocol: Leveraging Synthetic Data to Validate a Benchmark Study for Differential Abundance Tests for 16S Microbiome Sequencing Data.F1000Res. 2025 Jan 2;13:1180. doi: 10.12688/f1000research.155230.2. eCollection 2024. F1000Res. 2025. PMID: 39866725 Free PMC article.
-
Large-scale benchmarking reveals false discoveries and count transformation sensitivity in 16S rRNA gene amplicon data analysis methods used in microbiome studies.Microbiome. 2016 Nov 25;4(1):62. doi: 10.1186/s40168-016-0208-8. Microbiome. 2016. PMID: 27884206 Free PMC article.
-
Benchmarking Differential Abundance Tests for 16S microbiome sequencing data using simulated data based on experimental templates.PLoS One. 2025 May 19;20(5):e0321452. doi: 10.1371/journal.pone.0321452. eCollection 2025. PLoS One. 2025. PMID: 40388544 Free PMC article.
-
Identification of consensus head and neck cancer-associated microbiota signatures: a systematic review and meta-analysis of 16S rRNA and The Cancer Microbiome Atlas datasets.J Med Microbiol. 2024 Feb;73(2). doi: 10.1099/jmm.0.001799. J Med Microbiol. 2024. PMID: 38299619
-
Investigating differential abundance methods in microbiome data: A benchmark study.PLoS Comput Biol. 2022 Sep 8;18(9):e1010467. doi: 10.1371/journal.pcbi.1010467. eCollection 2022 Sep. PLoS Comput Biol. 2022. PMID: 36074761 Free PMC article. Review.
Cited by
-
Elementary methods provide more replicable results in microbial differential abundance analysis.Brief Bioinform. 2025 Mar 4;26(2):bbaf130. doi: 10.1093/bib/bbaf130. Brief Bioinform. 2025. PMID: 40135504 Free PMC article.
-
Computational Study Protocol: Leveraging Synthetic Data to Validate a Benchmark Study for Differential Abundance Tests for 16S Microbiome Sequencing Data.F1000Res. 2025 Jan 2;13:1180. doi: 10.12688/f1000research.155230.2. eCollection 2024. F1000Res. 2025. PMID: 39866725 Free PMC article.
-
Semisynthetic simulation for microbiome data analysis.Brief Bioinform. 2024 Nov 22;26(1):bbaf051. doi: 10.1093/bib/bbaf051. Brief Bioinform. 2024. PMID: 39927858 Free PMC article. Review.
-
Evaluation of extended-spectrum β-lactamase producing bacteria in feces of shelter dogs as a biomarker for altered gut microbial taxa and functional profiles.Front Microbiol. 2025 Mar 24;16:1556442. doi: 10.3389/fmicb.2025.1556442. eCollection 2025. Front Microbiol. 2025. PMID: 40196031 Free PMC article.
-
Consensus approach to differential abundance analysis detects few differences in the oral microbiome of pregnant women due to pre-existing type 2 diabetes mellitus.Microb Genom. 2025 Apr;11(4):001385. doi: 10.1099/mgen.0.001385. Microb Genom. 2025. PMID: 40232948 Free PMC article.
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
