

Exact Decoding of a Sequentially Markov Coalescent Model in Genetics
- PMID: 39323740
- PMCID: PMC11421421
- DOI: 10.1080/01621459.2023.2252570
Exact Decoding of a Sequentially Markov Coalescent Model in Genetics
Abstract
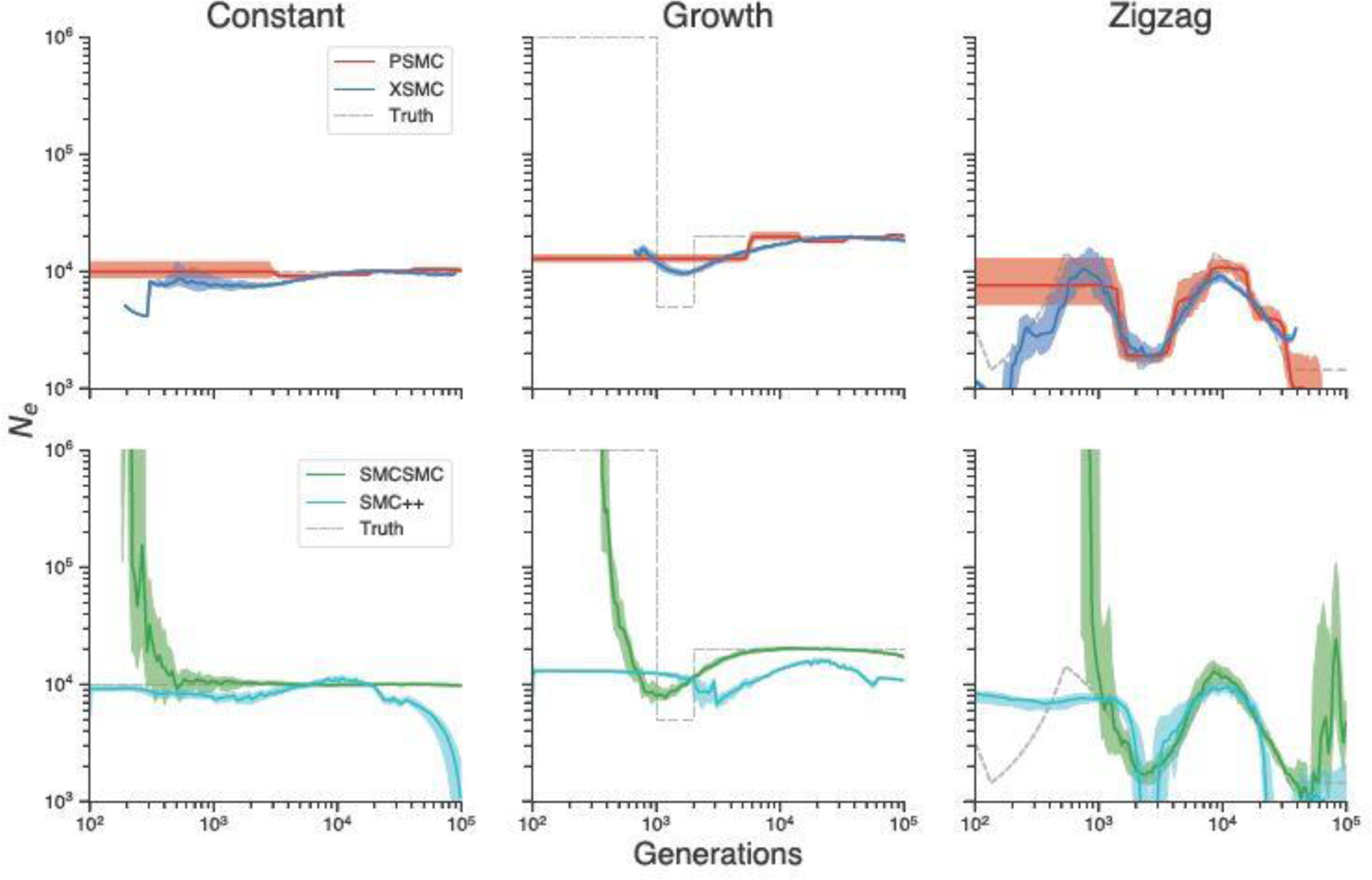
In statistical genetics, the sequentially Markov coalescent (SMC) is an important family of models for approximating the distribution of genetic variation data under complex evolutionary models. Methods based on SMC are widely used in genetics and evolutionary biology, with significant applications to genotype phasing and imputation, recombination rate estimation, and inferring population history. SMC allows for likelihood-based inference using hidden Markov models (HMMs), where the latent variable represents a genealogy. Because genealogies are continuous, while HMMs are discrete, SMC requires discretizing the space of trees in a way that is awkward and creates bias. In this work, we propose a method that circumvents this requirement, enabling SMC-based inference to be performed in the natural setting of a continuous state space. We derive fast, exact procedures for frequentist and Bayesian inference using SMC. Compared to existing methods, ours requires minimal user intervention or parameter tuning, no numerical optimization or E-M, and is faster and more accurate.
Keywords: changepoint; coalescent; hidden Markov model; population genetics.
Figures

Similar articles
-
Limits and convergence properties of the sequentially Markovian coalescent.Mol Ecol Resour. 2021 Oct;21(7):2231-2248. doi: 10.1111/1755-0998.13416. Epub 2021 May 30. Mol Ecol Resour. 2021. PMID: 33978324
-
Exact limits of inference in coalescent models.Theor Popul Biol. 2019 Feb;125:75-93. doi: 10.1016/j.tpb.2018.11.004. Epub 2018 Dec 17. Theor Popul Biol. 2019. PMID: 30571959 Free PMC article.
-
The Promise of Inferring the Past Using the Ancestral Recombination Graph.Genome Biol Evol. 2024 Feb 1;16(2):evae005. doi: 10.1093/gbe/evae005. Genome Biol Evol. 2024. PMID: 38242694 Free PMC article.
-
Phase-type distributions in mathematical population genetics: An emerging framework.Theor Popul Biol. 2024 Jun;157:14-32. doi: 10.1016/j.tpb.2024.03.001. Epub 2024 Mar 7. Theor Popul Biol. 2024. PMID: 38460602 Review.
-
Inference of population history using coalescent HMMs: review and outlook.Curr Opin Genet Dev. 2018 Dec;53:70-76. doi: 10.1016/j.gde.2018.07.002. Epub 2018 Jul 26. Curr Opin Genet Dev. 2018. PMID: 30056275 Free PMC article. Review.
Cited by
-
The solution surface of the Li-Stephens haplotype copying model.Algorithms Mol Biol. 2023 Aug 9;18(1):12. doi: 10.1186/s13015-023-00237-z. Algorithms Mol Biol. 2023. PMID: 37559098 Free PMC article.
-
Accelerated Bayesian inference of population size history from recombining sequence data.bioRxiv [Preprint]. 2024 Mar 27:2024.03.25.586640. doi: 10.1101/2024.03.25.586640. bioRxiv. 2024. PMID: 38585997 Free PMC article. Preprint.
References
-
- Adrion JR, Cole CB, Dukler N, Galloway JG, Gladstein AL, Gower G, Kyriazis CC, Ragsdale AP, Tsambos G, Baumdicker F, Carlson J, Cartwright RA, Durvasula A, Kim BY, McKenzie P, Messer PW, Noskova E, Vecchyo DO-D, Racimo F, Struck TJ, Gravel S, Gutenkunst RN, Lohmeuller KE, Ralph PL, Schrider DR, Siepel A, Kelleher J, and Kern AD (2019), “A community-maintained standard library of population genetic models,” bioRxiv,. - PMC - PubMed
-
- Barry D, and Hartigan JA (1992), “Product partition models for change point problems,” The Annals of Statistics, pp. 260–279.
-
- Barry D, and Hartigan JA (1993), “A Bayesian analysis for change point problems,” Journal of the American Statistical Association, 88(421), 309–319.
-
- Bishop CM (2006), Pattern Recognition and Machine Learning, Berlin, Heidelberg: Springer-Verlag.
Grants and funding
LinkOut - more resources
Full Text Sources