

TSC/mTORC1 mediates mTORC2/AKT1 signaling in c-MYC-induced murine hepatocarcinogenesis via centromere protein M
- PMID: 39325536
- PMCID: PMC11563669
- DOI: 10.1172/JCI174415
TSC/mTORC1 mediates mTORC2/AKT1 signaling in c-MYC-induced murine hepatocarcinogenesis via centromere protein M
Abstract
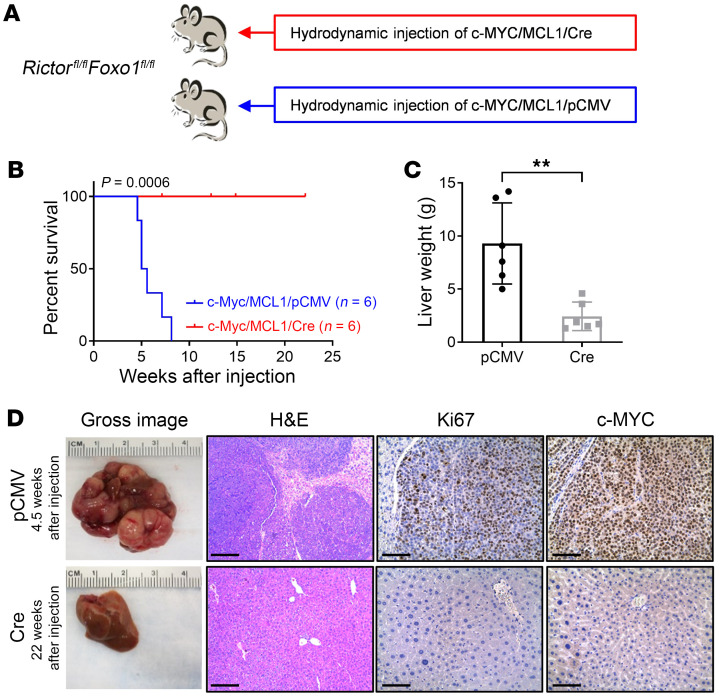
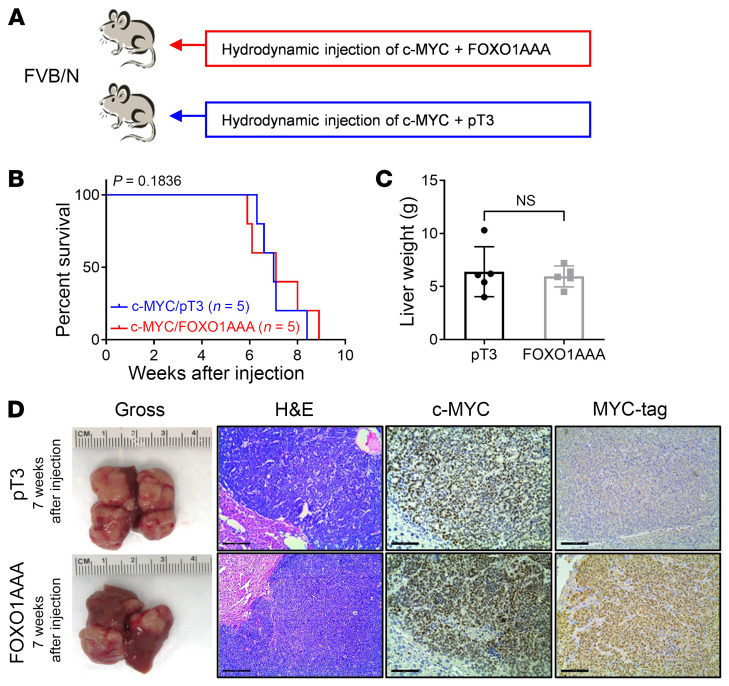
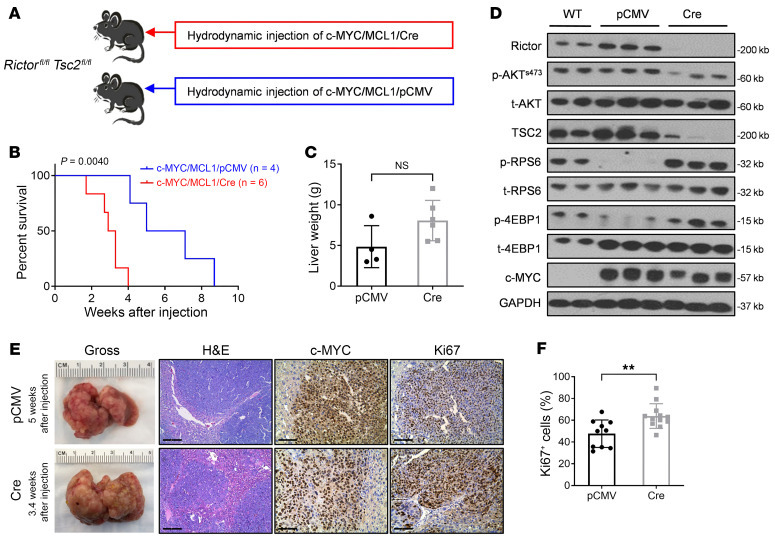
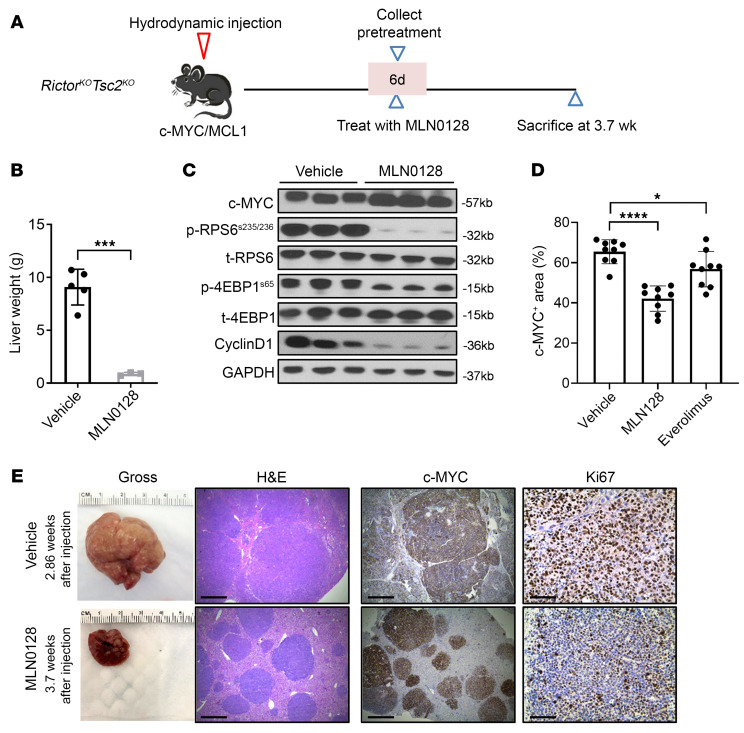
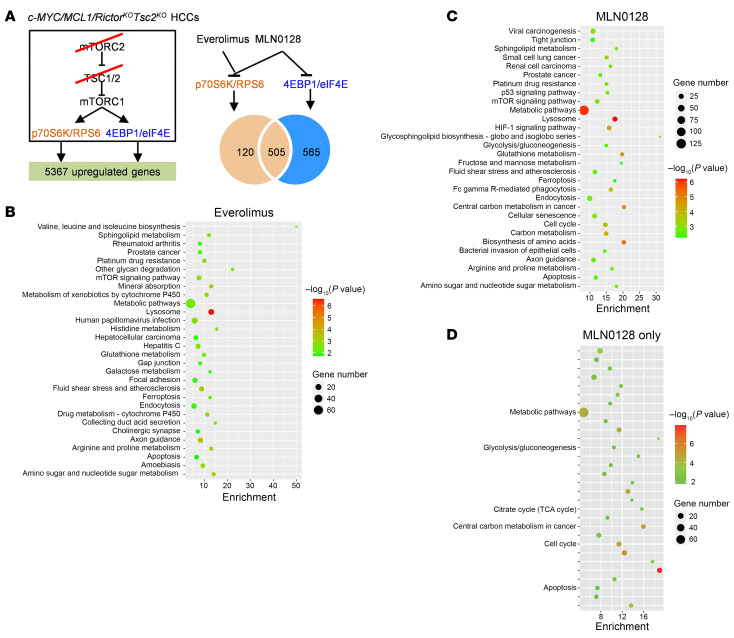
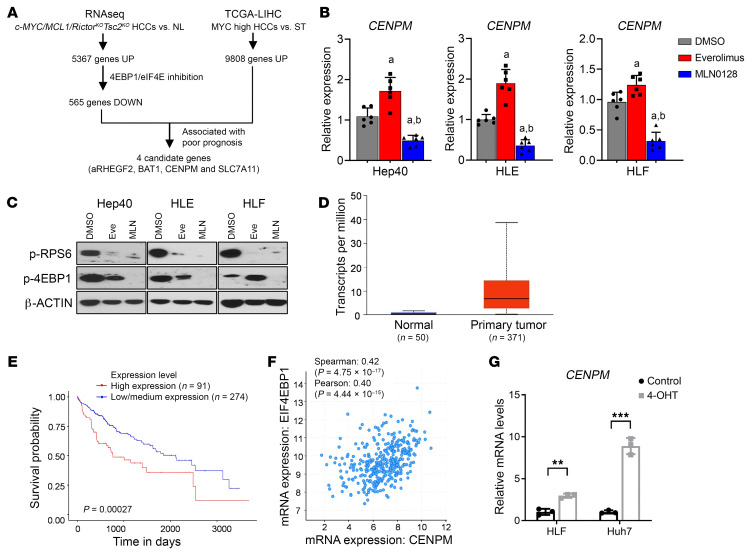
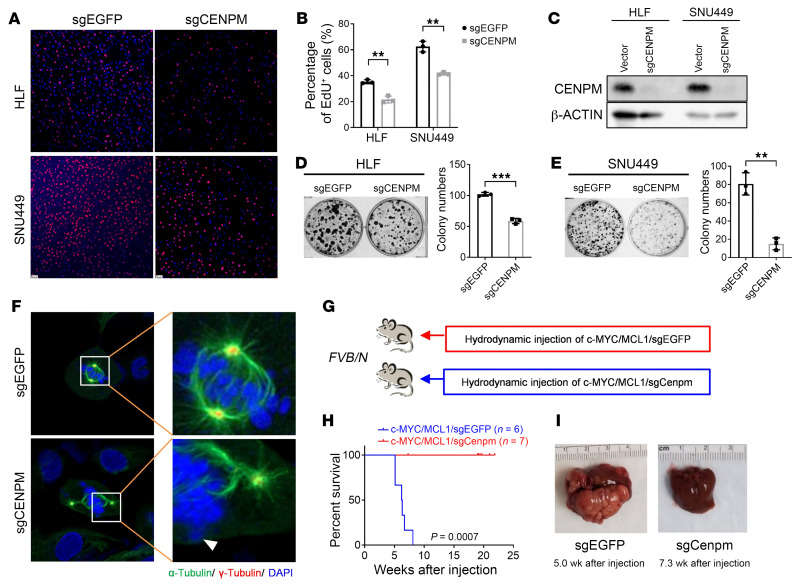
Activated mTORC2/AKT signaling plays a role in hepatocellular carcinoma (HCC). Research has shown that TSC/mTORC1 and FOXO1 are distinct downstream effectors of AKT signaling in liver regeneration and metabolism. However, the mechanisms by which these pathways mediate mTORC2/AKT activation in HCC are not yet fully understood. Amplification and activation of c-MYC are key molecular events in HCC. In this study, we explored the roles of tuberous sclerosis complex/mTORC1 (TSC/mTORC1) and FOXO1 as downstream effectors of mTORC2/AKT1 in c-MYC-induced hepatocarcinogenesis. Using various genetic approaches in mice, we found that manipulating the FOXO pathway had a minimal effect on c-MYC-induced HCC. In contrast, loss of mTORC2 inhibited c-MYC-induced HCC, an effect that was completely reversed by ablation of TSC2, which activated mTORC1. Additionally, we discovered that p70/RPS6 and 4EBP1/eIF4E acted downstream of mTORC1, regulating distinct molecular pathways. Notably, the 4EBP1/eIF4E cascade is crucial for cell proliferation and glycolysis in c-MYC-induced HCC. We also identified centromere protein M (CENPM) as a downstream target of the TSC2/mTORC1 pathway in c-MYC-driven hepatocarcinogenesis, and its ablation entirely inhibited c-MYC-dependent HCC formation. Our findings demonstrate that the TSC/mTORC1/CENPM pathway, rather than the FOXO cascade, is the primary signaling pathway regulating c-MYC-driven hepatocarcinogenesis. Targeting CENPM holds therapeutic potential for treating c-MYC-driven HCC.
Keywords: Hepatology; Liver cancer; Mouse models; Oncology; Signal transduction.
Figures
Similar articles
-
IGF2 Is Up-regulated by Epigenetic Mechanisms in Hepatocellular Carcinomas and Is an Actionable Oncogene Product in Experimental Models.Gastroenterology. 2016 Dec;151(6):1192-1205. doi: 10.1053/j.gastro.2016.09.001. Epub 2016 Sep 7. Gastroenterology. 2016. PMID: 27614046
-
mTORC1 Promotes ARID1A Degradation and Oncogenic Chromatin Remodeling in Hepatocellular Carcinoma.Cancer Res. 2021 Nov 15;81(22):5652-5665. doi: 10.1158/0008-5472.CAN-21-0206. Epub 2021 Aug 24. Cancer Res. 2021. PMID: 34429326 Free PMC article.
-
4EBP1/eIF4E and p70S6K/RPS6 axes play critical and distinct roles in hepatocarcinogenesis driven by AKT and N-Ras proto-oncogenes in mice.Hepatology. 2015 Jan;61(1):200-13. doi: 10.1002/hep.27396. Epub 2014 Nov 25. Hepatology. 2015. PMID: 25145583 Free PMC article.
-
The Spectrum of Renal "TFEopathies": Flipping the mTOR Switch in Renal Tumorigenesis.Physiology (Bethesda). 2024 Nov 1;39(6):0. doi: 10.1152/physiol.00026.2024. Epub 2024 Jul 16. Physiology (Bethesda). 2024. PMID: 39012319 Review.
-
Evolution of growth factor signaling to the TSC complex to regulate mTORC1.Sci Signal. 2025 Jul 8;18(894):eadw4165. doi: 10.1126/scisignal.adw4165. Epub 2025 Jul 8. Sci Signal. 2025. PMID: 40627686 Review.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
