

ERK hyperactivation in epidermal keratinocytes impairs intercellular adhesion and drives Grover disease pathology
- PMID: 39325541
- PMCID: PMC11601706
- DOI: 10.1172/jci.insight.182983
ERK hyperactivation in epidermal keratinocytes impairs intercellular adhesion and drives Grover disease pathology
Abstract
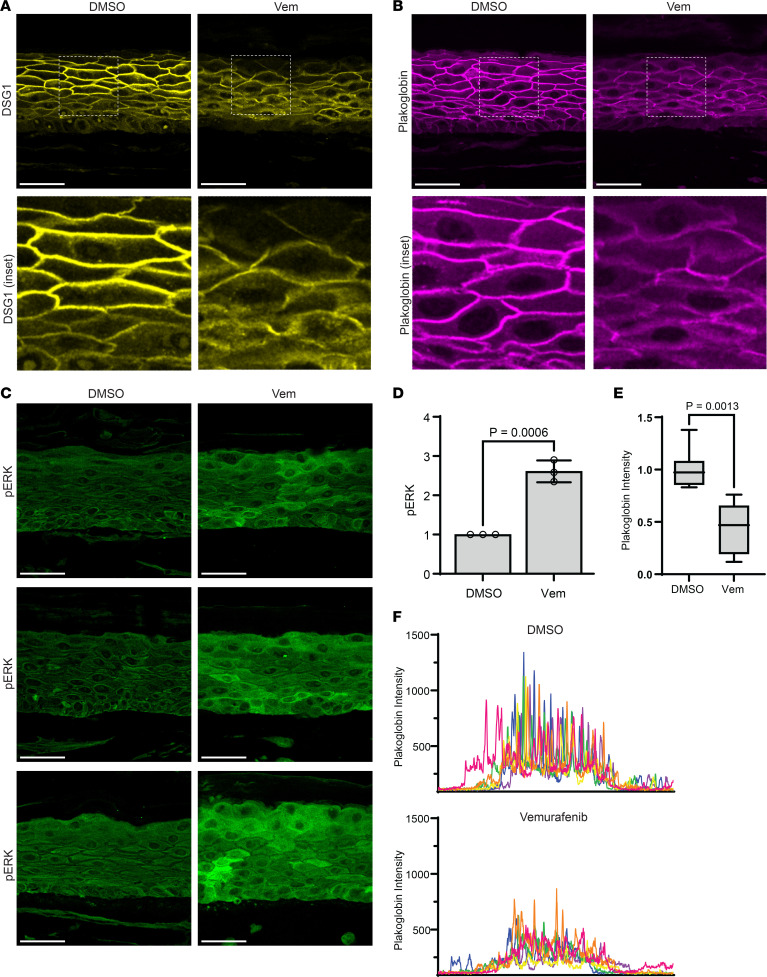
Grover disease is an acquired epidermal blistering disorder in which keratinocytes lose intercellular connections. While its pathologic features are well defined, its etiology remains unclear, and there is no FDA-approved therapy. Interestingly, Grover disease was a common adverse event in clinical trials for cancer using B-RAF inhibitors, but it remained unknown how B-RAF blockade compromised skin integrity. Here, we identified ERK hyperactivation as a key driver of Grover disease pathology. We leveraged a fluorescent biosensor to confirm that the B-RAF inhibitors dabrafenib and vemurafenib paradoxically activated ERK in human keratinocytes and organotypic epidermis, disrupting cell-cell junctions and weakening epithelial integrity. Consistent with clinical data showing that concomitant MEK blockade prevents Grover disease in patients receiving B-RAF inhibitors, we found that MEK inhibition suppressed ERK and rescued cohesion of B-RAF-inhibited keratinocytes. Validating these results, we demonstrated ERK hyperactivation in patient biopsies from vemurafenib-induced Grover disease and from spontaneous Grover disease, revealing a common etiology for both. Finally, in line with our recent identification of ERK hyperactivation in Darier disease, a genetic disorder with identical pathology to Grover disease, our studies uncovered that the pathogenic mechanisms of these diseases converge on ERK signaling and support MEK inhibition as a therapeutic strategy.
Keywords: Cell biology; Cell migration/adhesion; Dermatology; Signal transduction; Skin.
Figures





Update of
-
ERK hyperactivation in epidermal keratinocytes impairs intercellular adhesion and drives Grover disease pathology.bioRxiv [Preprint]. 2024 May 3:2024.04.30.591953. doi: 10.1101/2024.04.30.591953. bioRxiv. 2024. Update in: JCI Insight. 2024 Nov 8;9(21):e182983. doi: 10.1172/jci.insight.182983. PMID: 38746263 Free PMC article. Updated. Preprint.
References
-
- Karray M, et al. Kaposi Varicelliform Eruption. In: Ackley WB, et al, eds. StatPearls. Treasure Island; 2022.
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
