

Human cells contain myriad excised linear intron RNAs with links to gene regulation and potential utility as biomarkers
- PMID: 39325823
- PMCID: PMC11460701
- DOI: 10.1371/journal.pgen.1011416
Human cells contain myriad excised linear intron RNAs with links to gene regulation and potential utility as biomarkers
Abstract
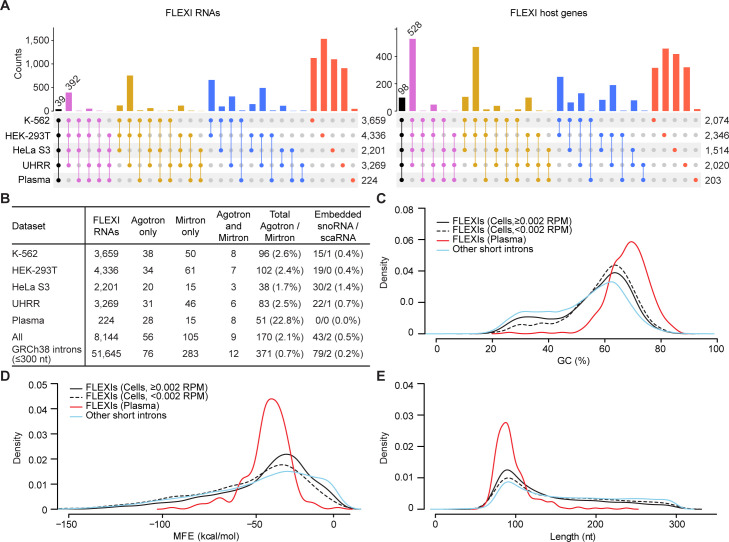
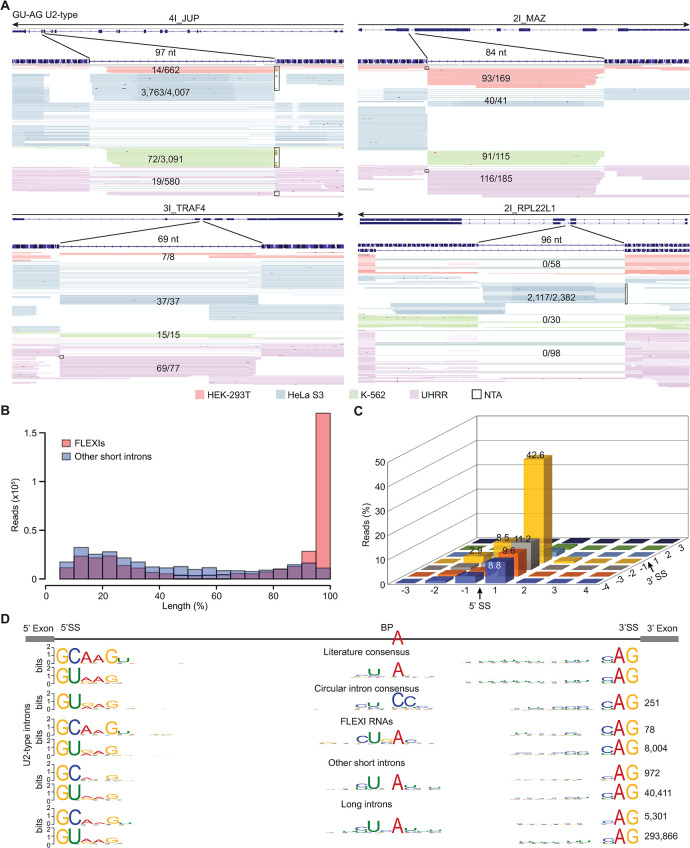
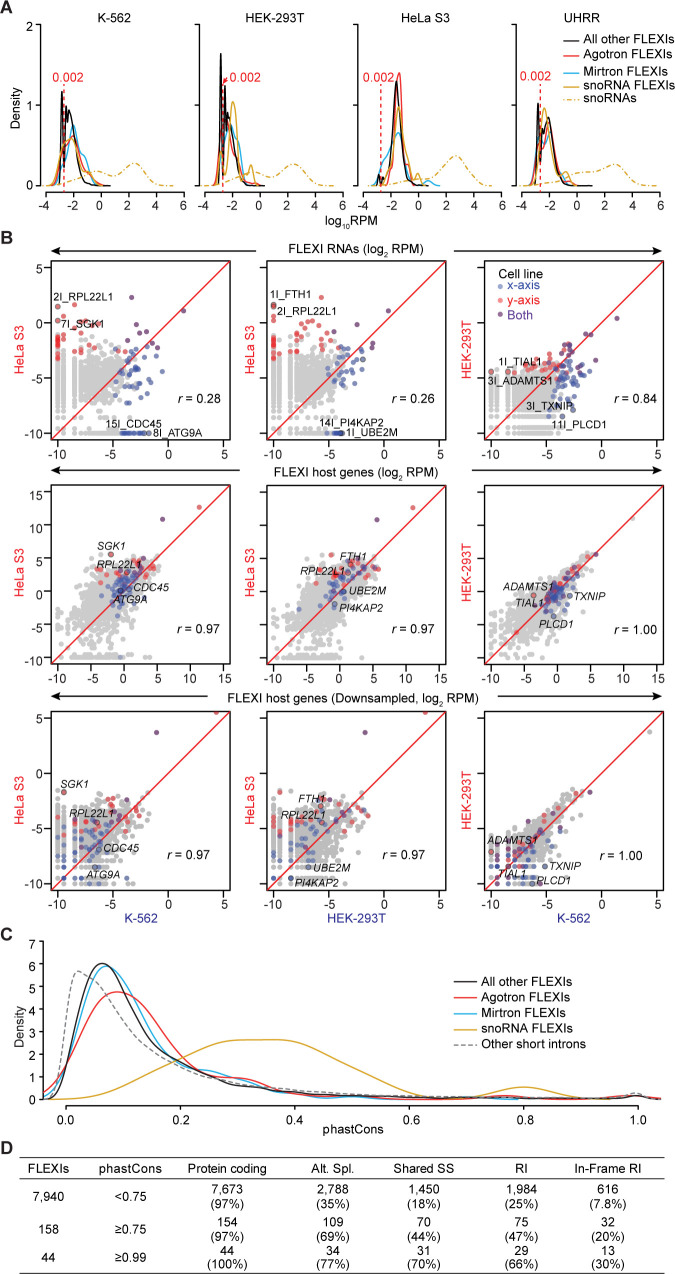
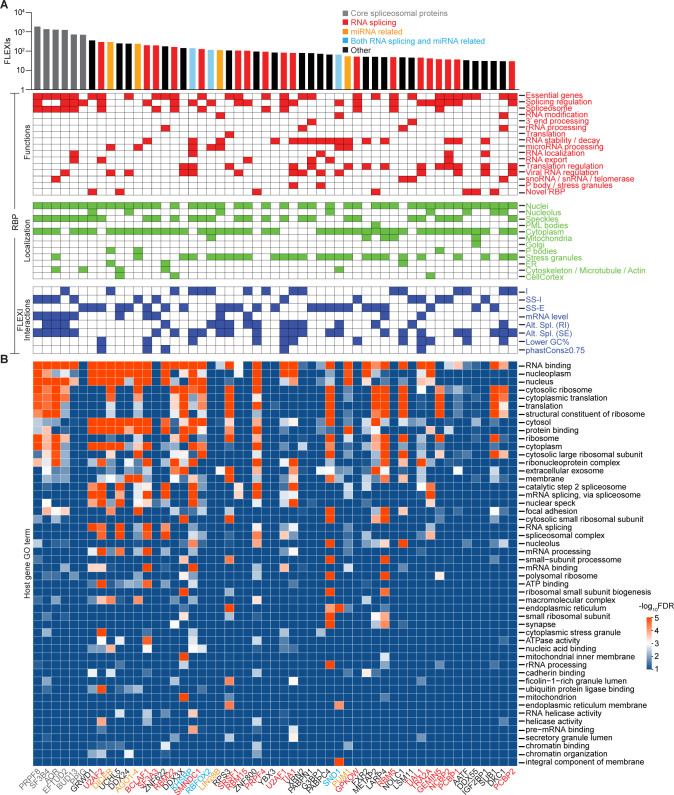
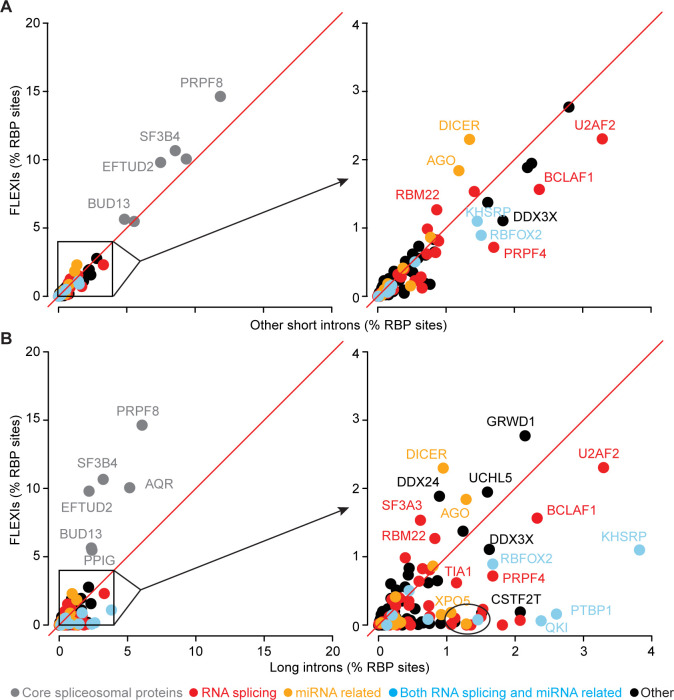
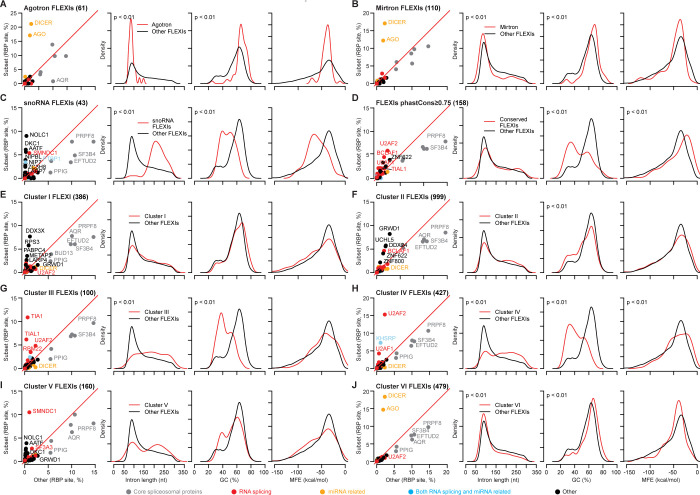
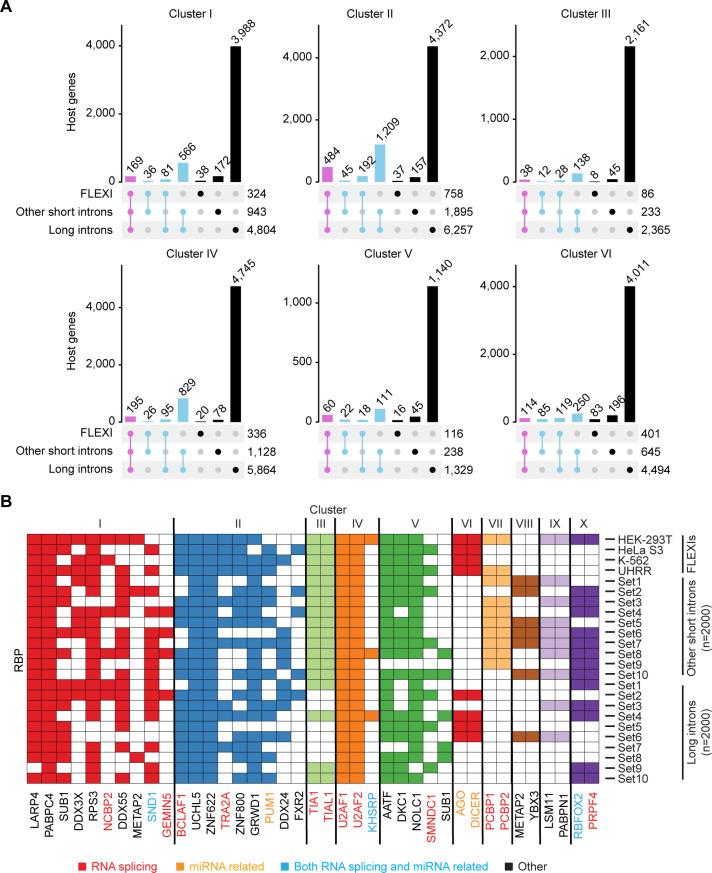
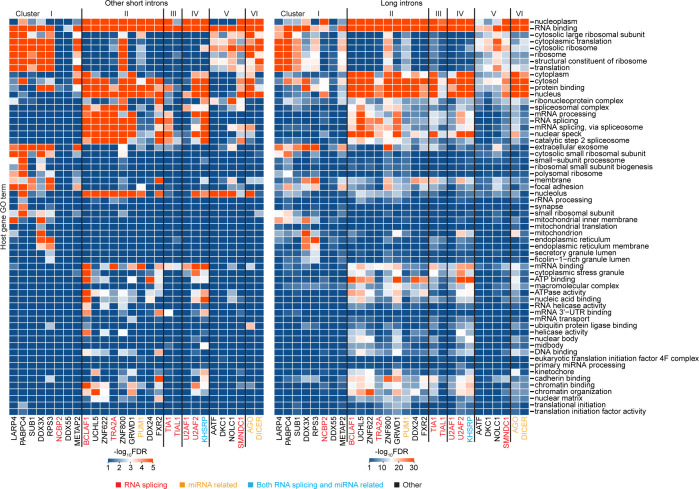
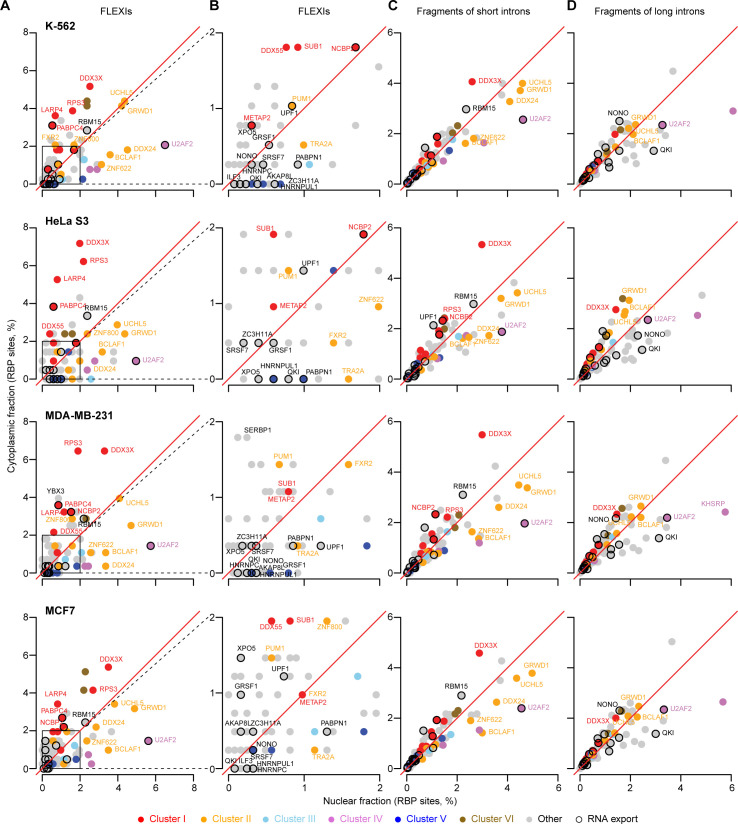
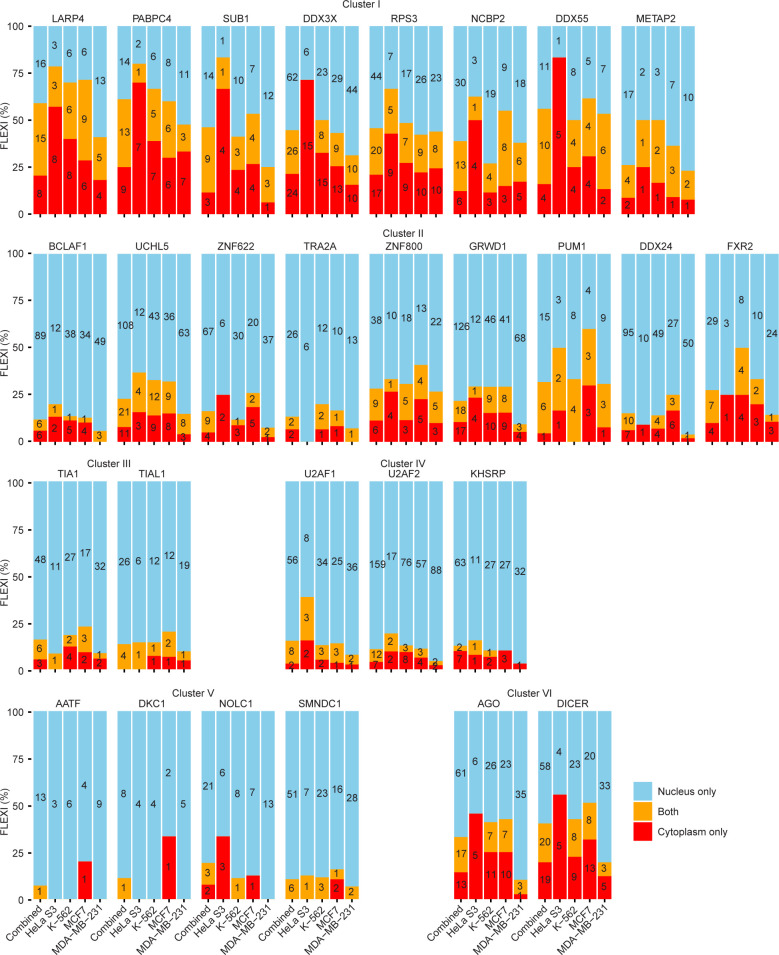
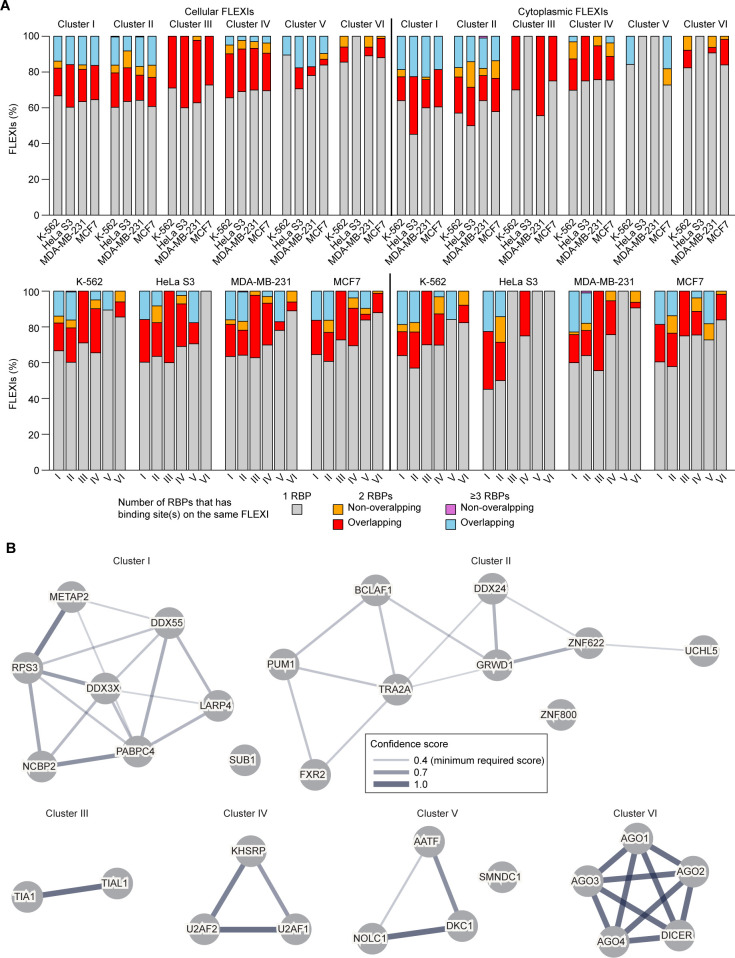
A previous study using Thermostable Group II Intron Reverse Transcriptase sequencing (TGIRT-seq) found human plasma contains short (≤300 nt) structured full-length excised linear intron (FLEXI) RNAs with potential to serve as blood-based biomarkers. Here, TGIRT-seq identified >9,000 different FLEXI RNAs in human cell lines, including relatively abundant FLEXIs with cell-type-specific expression patterns. Analysis of public CLIP-seq datasets identified 126 RNA-binding proteins (RBPs) that have binding sites within the region corresponding to the FLEXI or overlapping FLEXI splice sites in pre-mRNAs, including 53 RBPs with binding sites for ≥30 different FLEXIs. These included splicing factors, transcription factors, a chromatin remodeling protein, cellular growth regulators, and proteins with cytoplasmic functions. Analysis of ENCODE datasets identified subsets of these RBPs whose knockdown impacted FLEXI host gene mRNA levels or proximate alternative splicing, indicating functional interactions. Hierarchical clustering identified six subsets of RBPs whose FLEXI binding sites were co-enriched in six subsets of functionally related host genes: AGO1-4 and DICER, including but not limited to agotrons or mirtron pre-miRNAs; DKC1, NOLC1, SMNDC1, and AATF (Apoptosis Antagonizing Transcription Factor), including but not limited to snoRNA-encoding FLEXIs; two subsets of alternative splicing factors; and two subsets that included RBPs with cytoplasmic functions (e.g., LARP4, PABPC4, METAP2, and ZNF622) together with regulatory proteins. Cell fractionation experiments showed cytoplasmic enrichment of FLEXI RNAs with binding sites for RBPs with cytoplasmic functions. The subsets of host genes encoding FLEXIs with binding sites for different subsets of RBPs were co-enriched with non-FLEXI other short and long introns with binding sites for the same RBPs, suggesting overarching mechanisms for coordinately regulating expression of functionally related genes. Our findings identify FLEXIs as a previously unrecognized large class of cellular RNAs and provide a comprehensive roadmap for further analyzing their biological functions and the relationship of their RBPs to cellular regulatory mechanisms.
Copyright: © 2024 Yao et al. This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Conflict of interest statement
I have read the journal’s policy and the authors of this manuscript have the following competing interests: AML is an inventor on patents owned by the University of Texas at Austin for TGIRT enzymes and other stabilized reverse transcriptase fusion proteins and methods for non-retroviral reverse transcriptase template switching. AML, JY, and HX are inventors on a patent application filed by the University of Texas for the use of FLEXIs and other intron RNAs as biomarkers.
Figures

Similar articles
-
Identification of protein-protected mRNA fragments and structured excised intron RNAs in human plasma by TGIRT-seq peak calling.Elife. 2020 Sep 2;9:e60743. doi: 10.7554/eLife.60743. Elife. 2020. PMID: 32876046 Free PMC article.
-
TGIRT-seq Protocol for the Comprehensive Profiling of Coding and Non-coding RNA Biotypes in Cellular, Extracellular Vesicle, and Plasma RNAs.Bio Protoc. 2021 Dec 5;11(23):e4239. doi: 10.21769/BioProtoc.4239. eCollection 2021 Dec 5. Bio Protoc. 2021. PMID: 35005084 Free PMC article.
-
Long Non-Coding RNA Expression Levels Modulate Cell-Type-Specific Splicing Patterns by Altering Their Interaction Landscape with RNA-Binding Proteins.Genes (Basel). 2019 Aug 6;10(8):593. doi: 10.3390/genes10080593. Genes (Basel). 2019. PMID: 31390792 Free PMC article.
-
New sequencing methodologies reveal interplay between multiple RNA-binding proteins and their RNAs.Curr Genet. 2020 Aug;66(4):713-717. doi: 10.1007/s00294-020-01066-y. Epub 2020 Mar 19. Curr Genet. 2020. PMID: 32193580 Free PMC article. Review.
-
Understanding splicing regulation through RNA splicing maps.Trends Genet. 2011 Mar;27(3):89-97. doi: 10.1016/j.tig.2010.12.001. Epub 2011 Jan 12. Trends Genet. 2011. PMID: 21232811 Free PMC article. Review.
Cited by
-
Intron-lariat spliceosomes convert lariats to true circles: implications for intron transposition.bioRxiv [Preprint]. 2024 Mar 27:2024.03.26.586863. doi: 10.1101/2024.03.26.586863. bioRxiv. 2024. Update in: Genes Dev. 2024 May 21;38(7-8):322-335. doi: 10.1101/gad.351764.124. PMID: 38585890 Free PMC article. Updated. Preprint.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
