

Activating mutations remodel the chromatin accessibility landscape to drive distinct regulatory networks in KMT2A-rearranged acute leukemia
- PMID: 39329074
- PMCID: PMC11426354
- DOI: 10.1002/hem3.70006
Activating mutations remodel the chromatin accessibility landscape to drive distinct regulatory networks in KMT2A-rearranged acute leukemia
Abstract
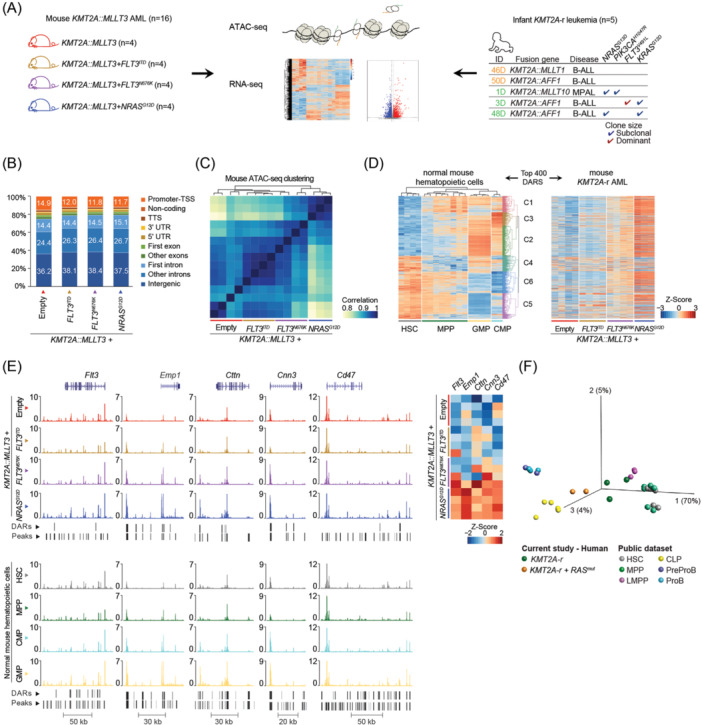
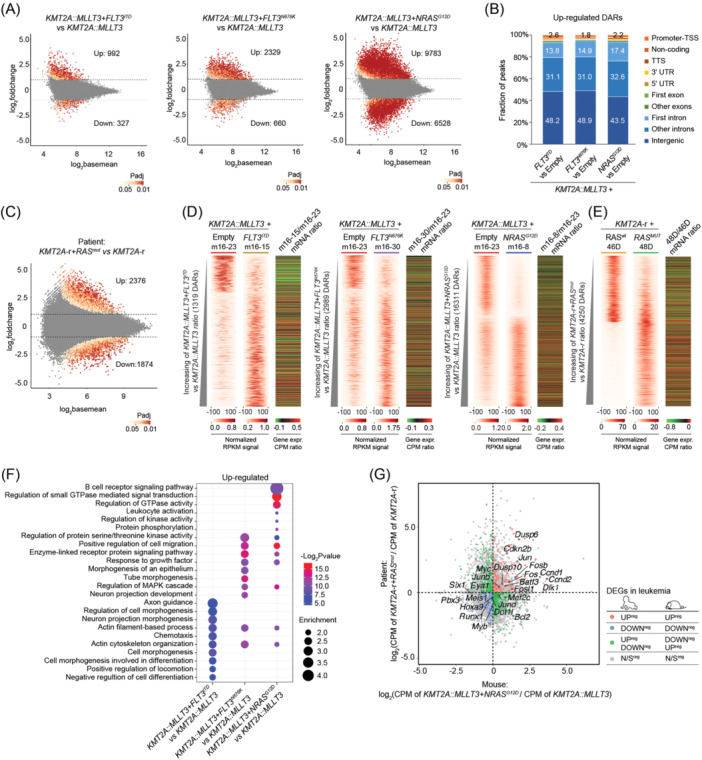
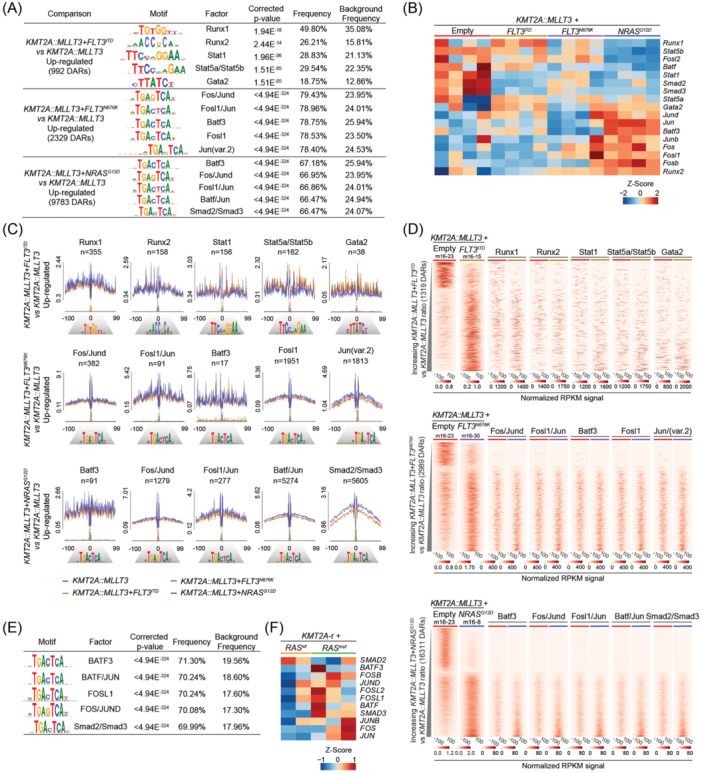
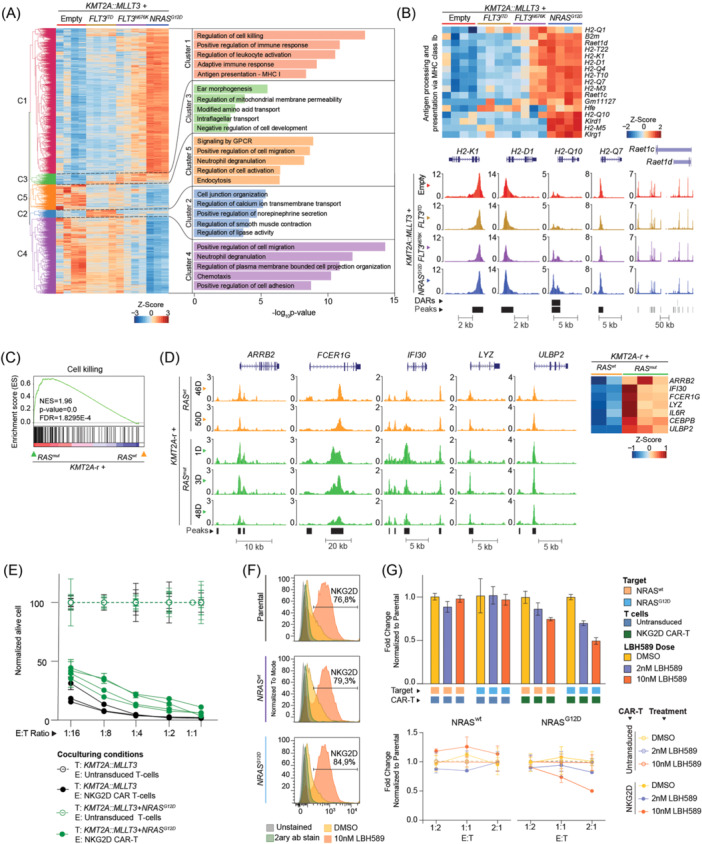
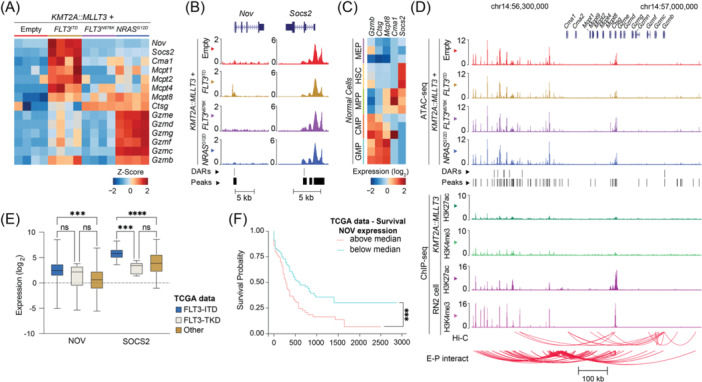
Activating FLT3 and RAS mutations commonly occur in leukemia with KMT2A-gene rearrangements (KMT2A-r). However, how these mutations cooperate with the KMT2A-r to remodel the epigenetic landscape is unknown. Using a retroviral acute myeloid leukemia (AML) mouse model driven by KMT2A::MLLT3, we show that FLT3 ITD , FLT3 N676K , and NRAS G12D remodeled the chromatin accessibility landscape and associated transcriptional networks. Although the activating mutations shared a common core of chromatin changes, each mutation exhibits unique profiles with most opened peaks associating with enhancers in intronic or intergenic regions. Specifically, FLT3 N676K and NRAS G12D rewired similar chromatin and transcriptional networks, distinct from those mediated by FLT3 ITD . Motif analysis uncovered a role for the AP-1 family of transcription factors in KMT2A::MLLT3 leukemia with FLT3 N676K and NRAS G12D , whereas Runx1 and Stat5a/Stat5b were active in the presence of FLT3 ITD . Furthermore, transcriptional programs linked to immune cell regulation were activated in KMT2A-r AML expressing NRAS G12D or FLT3 N676K , and the expression of NKG2D-ligands on KMT2A-r cells rendered them sensitive to CAR T cell-mediated killing. Human KMT2A-r AML cells could be pharmacologically sensitized to NKG2D-CAR T cells by treatment with the histone deacetylase inhibitor LBH589 (panobinostat) which caused upregulation of NKG2D-ligand levels. Co-treatment with LBH589 and NKG2D-CAR T cells enabled robust AML cell killing, and the strongest effect was observed for cells expressing NRAS G12D . Finally, the results were validated and extended to acute leukemia in infancy. Combined, activating mutations induced mutation-specific changes in the epigenetic landscape, leading to changes in transcriptional programs orchestrated by specific transcription factor networks.
© 2024 The Author(s). HemaSphere published by John Wiley & Sons Ltd on behalf of European Hematology Association.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Pieters R, De Lorenzo P, Ancliffe P, et al. Outcome of infants younger than 1 year with acute lymphoblastic leukemia treated with the interfant‐06 protocol: results from an international phase III randomized study. J Clin Oncol. 2019;37(25):2246‐2256. - PubMed
-
- van der Sluis IM, de Lorenzo P, Kotecha RS, et al. Blinatumomab added to chemotherapy in infant lymphoblastic leukemia. N Engl J Med. 2023;388(17):1572‐1581. - PubMed
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous