

Abnormal synaptic architecture in iPSC-derived neurons from a multi-generational family with genetic Creutzfeldt-Jakob disease
- PMID: 39332406
- PMCID: PMC11561462
- DOI: 10.1016/j.stemcr.2024.08.010
Abnormal synaptic architecture in iPSC-derived neurons from a multi-generational family with genetic Creutzfeldt-Jakob disease
Abstract
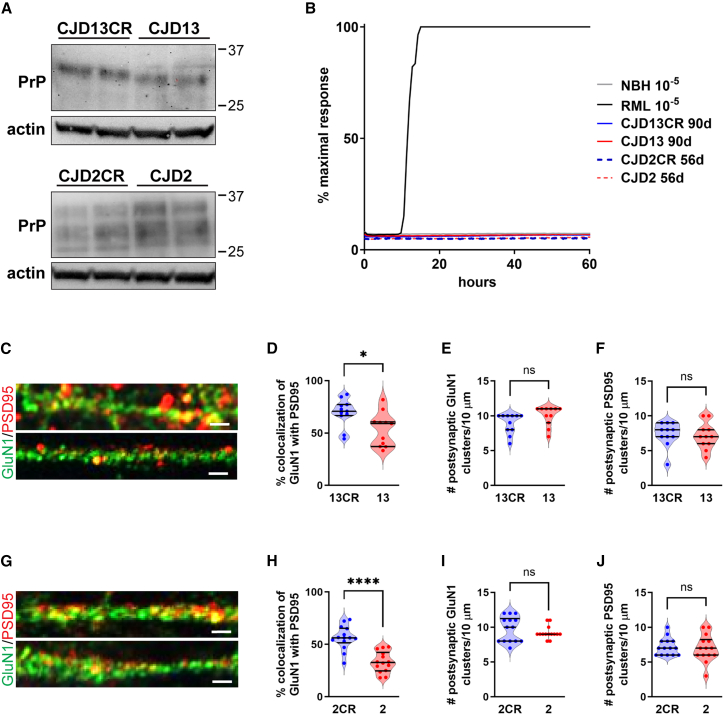
Genetic prion diseases are caused by mutations in PRNP, which encodes the prion protein (PrPC). Why these mutations are pathogenic, and how they alter the properties of PrPC are poorly understood. We have consented and accessed 22 individuals of a multi-generational Israeli family harboring the highly penetrant E200K PRNP mutation and generated a library of induced pluripotent stem cells (iPSCs) representing nine carriers and four non-carriers. iPSC-derived neurons from E200K carriers display abnormal synaptic architecture characterized by misalignment of postsynaptic NMDA receptors with the cytoplasmic scaffolding protein PSD95. Differentiated neurons from mutation carriers do not produce PrPSc, the aggregated and infectious conformer of PrP, suggesting that loss of a physiological function of PrPC may contribute to the disease phenotype. Our study shows that iPSC-derived neurons can provide important mechanistic insights into the pathogenesis of genetic prion diseases and can offer a powerful platform for testing candidate therapeutics.
Keywords: E200K; PrP; PrPSc; genetic prion disease; iPSC; neurodegeneration; prion; prion disease; synapse.
Copyright © 2024 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures





References
-
- Adle-Biassette H., Verney C., Peoc'h K., Dauge M.-C., Razavi F., Choudat L., Gressens P., Budka H., Henin D. Immunohistochemical expression of prion protein (PrPC) in the human forebrain during development. J. Neuropathol. Exp. Neurol. 2006;65:698–706. - PubMed
-
- Asante E.A., Gowland I., Grimshaw A., Linehan J.M., Smidak M., Houghton R., Osiguwa O., Tomlinson A., Joiner S., Brandner S., et al. Absence of spontaneous disease and comparative prion susceptibility of transgenic mice expressing mutant human prion proteins. J. Gen. Virol. 2009;90:546–558. - PMC - PubMed
-
- Asante E.A., Linehan J.M., Smidak M., Tomlinson A., Grimshaw A., Jeelani A., Jakubcova T., Hamdan S., Powell C., Brandner S., et al. Inherited prion disease A117V is not simply a proteinopathy but produces prions transmissible to transgenic mice expressing homologous prion protein. PLoS Pathog. 2013;9:e1003643. - PMC - PubMed
-
- Biasini E., Unterberger U., Solomon I.H., Massignan T., Senatore A., Bian H., Voigtlaender T., Bowman F.P., Bonetto V., Chiesa R., et al. A mutant prion protein sensitizes neurons to glutamate-induced excitotoxicity. J. Neurosci. 2013;33:2408–2418. doi: 10.1523/JNEUROSCI.3406-12.2013. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
