

The pleiotropic spectrum of proximal 16p11.2 CNVs
- PMID: 39332410
- PMCID: PMC11568765
- DOI: 10.1016/j.ajhg.2024.08.015
The pleiotropic spectrum of proximal 16p11.2 CNVs
Abstract
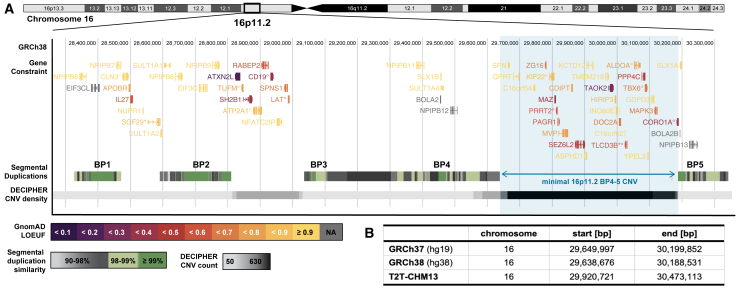
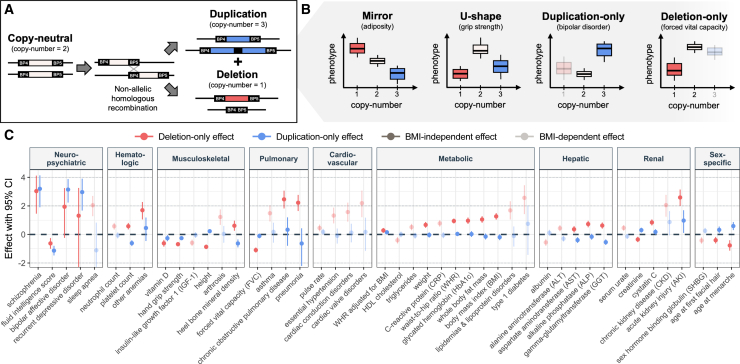
Recurrent genomic rearrangements at 16p11.2 BP4-5 represent one of the most common causes of genomic disorders. Originally associated with increased risk for autism spectrum disorder, schizophrenia, and intellectual disability, as well as adiposity and head circumference, these CNVs have since been associated with a plethora of phenotypic alterations, albeit with high variability in expressivity and incomplete penetrance. Here, we comprehensively review the pleiotropy associated with 16p11.2 BP4-5 rearrangements to shine light on its full phenotypic spectrum. Illustrating this phenotypic heterogeneity, we expose many parallels between findings gathered from clinical versus population-based cohorts, which often point to the same physiological systems, and emphasize the role of the CNV beyond neuropsychiatric and anthropometric traits. Revealing the complex and variable clinical manifestations of this CNV is crucial for accurate diagnosis and personalized treatment strategies for carrier individuals. Furthermore, we discuss areas of research that will be key to identifying factors contributing to phenotypic heterogeneity and gaining mechanistic insights into the molecular pathways underlying observed associations, while demonstrating how diversity in affected individuals, cohorts, experimental models, and analytical approaches can catalyze discoveries.
Keywords: multi-system disorder; penetrance; pleiotropy; proximal 16p11.2 BP4-5 CNV; structural variant; variable expressivity.
Copyright © 2024 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures



References
-
- Nuttle X., Giannuzzi G., Duyzend M.H., Schraiber J.G., Narvaiza I., Sudmant P.H., Penn O., Chiatante G., Malig M., Huddleston J., et al. Emergence of a Homo sapiens-specific gene family and chromosome 16p11.2 CNV susceptibility. Nature. 2016;536:205–209. doi: 10.1038/nature19075. - DOI - PMC - PubMed
-
- Migliavacca E., Golzio C., Männik K., Blumenthal I., Oh E.C., Harewood L., Kosmicki J.A., Loviglio M.N., Giannuzzi G., Hippolyte L., et al. A Potential Contributory Role for Ciliary Dysfunction in the 16p11.2 600 kb BP4-BP5 Pathology. Am. J. Hum. Genet. 2015;96:784–796. doi: 10.1016/j.ajhg.2015.04.002. - DOI - PMC - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
