

Identifying targetable metabolic dependencies across colorectal cancer progression
- PMID: 39332495
- PMCID: PMC11490841
- DOI: 10.1016/j.molmet.2024.102037
Identifying targetable metabolic dependencies across colorectal cancer progression
Abstract
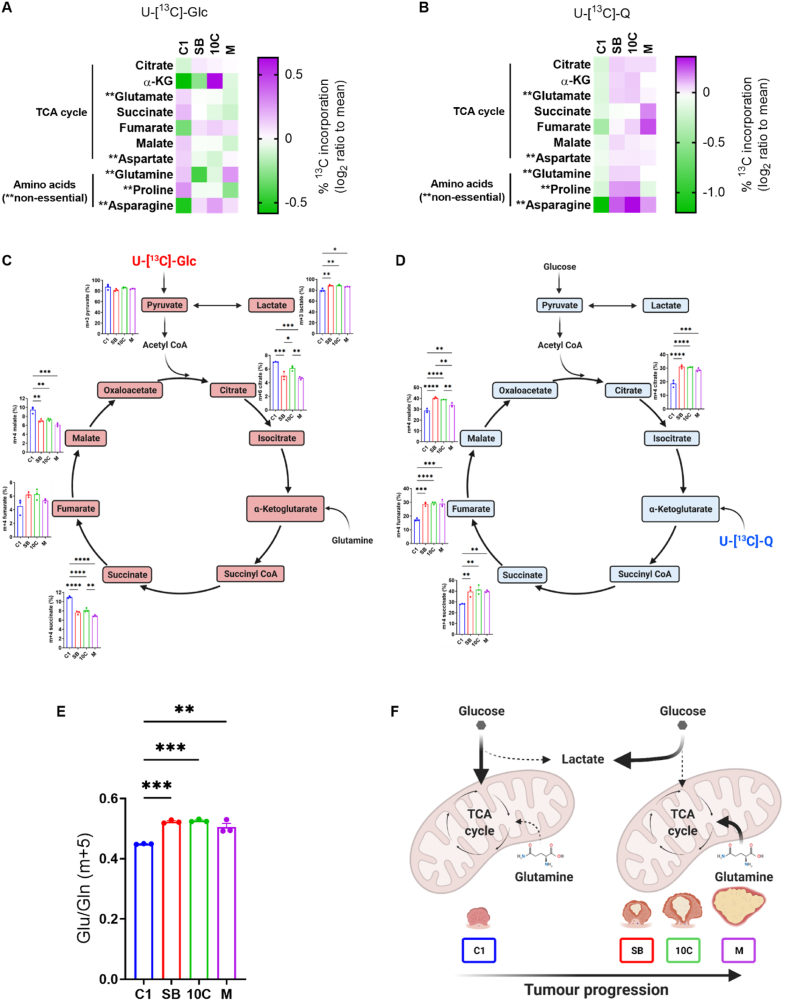
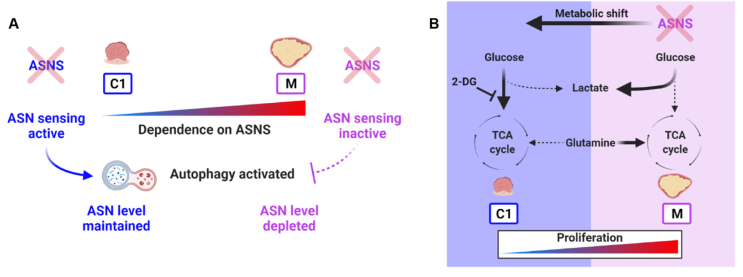
Colorectal cancer (CRC) is a multi-stage process initiated through the formation of a benign adenoma, progressing to an invasive carcinoma and finally metastatic spread. Tumour cells must adapt their metabolism to support the energetic and biosynthetic demands associated with disease progression. As such, targeting cancer cell metabolism is a promising therapeutic avenue in CRC. However, to identify tractable nodes of metabolic vulnerability specific to CRC stage, we must understand how metabolism changes during CRC development. Here, we use a unique model system - comprising human early adenoma to late adenocarcinoma. We show that adenoma cells transition to elevated glycolysis at the early stages of tumour progression but maintain oxidative metabolism. Progressed adenocarcinoma cells rely more on glutamine-derived carbon to fuel the TCA cycle, whereas glycolysis and TCA cycle activity remain tightly coupled in early adenoma cells. Adenocarcinoma cells are more flexible with respect to fuel source, enabling them to proliferate in nutrient-poor environments. Despite this plasticity, we identify asparagine (ASN) synthesis as a node of metabolic vulnerability in late-stage adenocarcinoma cells. We show that loss of asparagine synthetase (ASNS) blocks their proliferation, whereas early adenoma cells are largely resistant to ASN deprivation. Mechanistically, we show that late-stage adenocarcinoma cells are dependent on ASNS to support mTORC1 signalling and maximal glycolytic and oxidative capacity. Resistance to ASNS loss in early adenoma cells is likely due to a feedback loop, absent in late-stage cells, allowing them to sense and regulate ASN levels and supplement ASN by autophagy. Together, our study defines metabolic changes during CRC development and highlights ASN synthesis as a targetable metabolic vulnerability in later stage disease.
Keywords: Adenocarcinoma; Adenoma; Asparagine; Asparagine synthetase; Colorectal cancer; Oncometabolism.
Copyright © 2024 The Authors. Published by Elsevier GmbH.. All rights reserved.
Conflict of interest statement
Declaration of competing interest The authors declare that they have no conflict of interest.
Figures





References
-
- Sung H., Ferlay J., Siegel R.L., Laversanne M., Soerjomataram I., Jemal A., et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA A Cancer J Clin. 2021;71(3):209–249. - PubMed
-
- Fearon E.R., Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61(5):759–767. - PubMed
-
- Jackman R.J., Mayo C.W. The adenoma-carcinoma sequence in cancer of the colon. Surg Gynecol Obstet. 1951;93(3):327–330. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
