

A cell type-aware framework for nominating non-coding variants in Mendelian regulatory disorders
- PMID: 39333082
- PMCID: PMC11436875
- DOI: 10.1038/s41467-024-52463-7
A cell type-aware framework for nominating non-coding variants in Mendelian regulatory disorders
Abstract
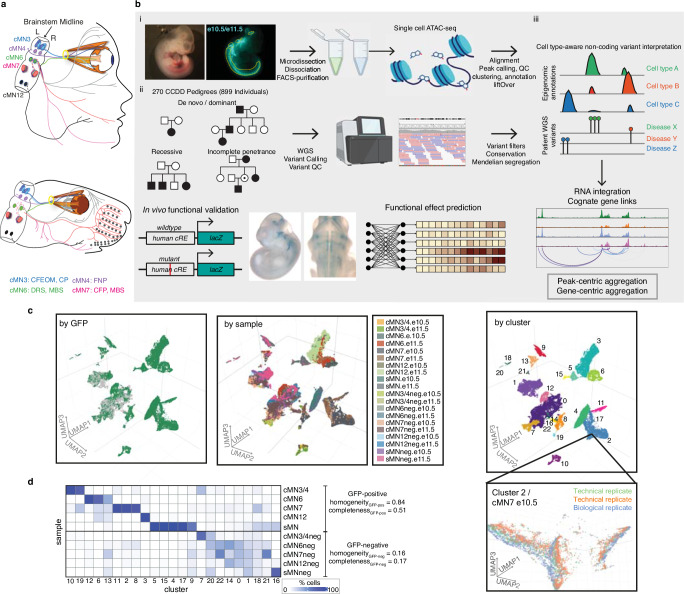
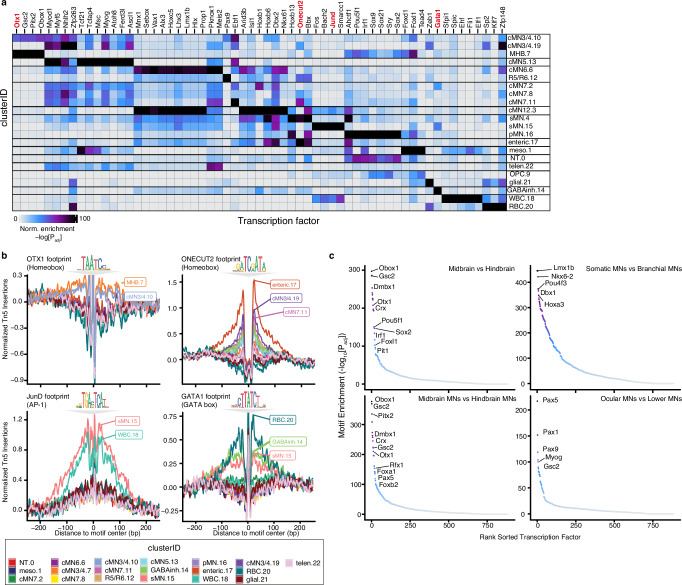
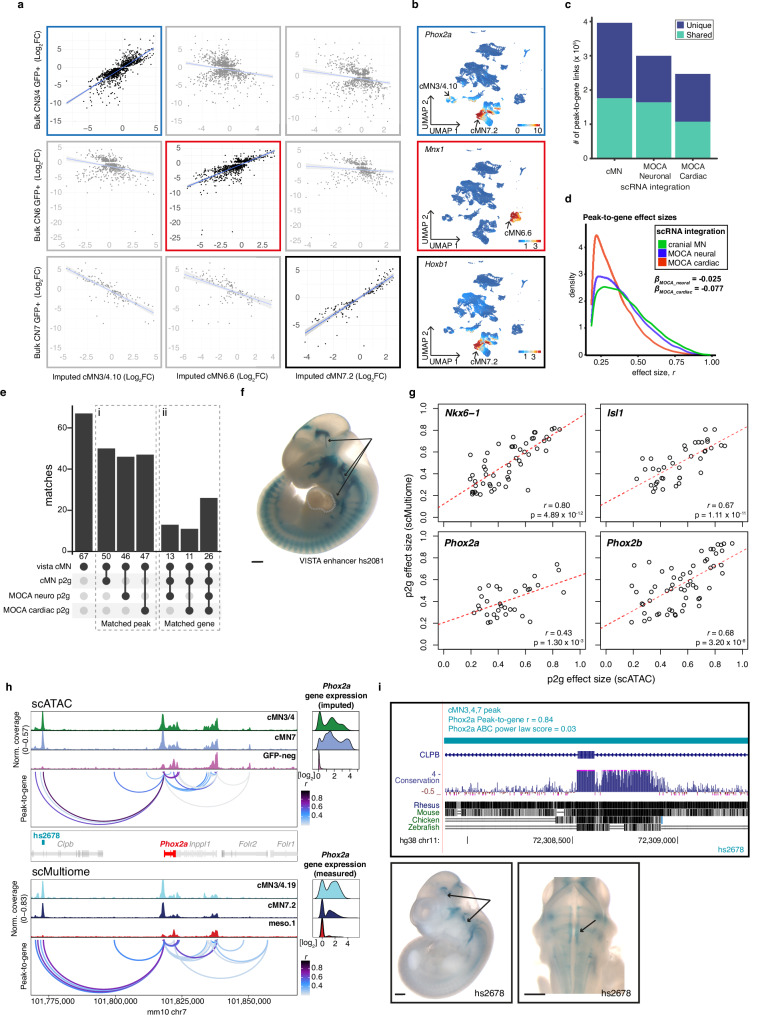
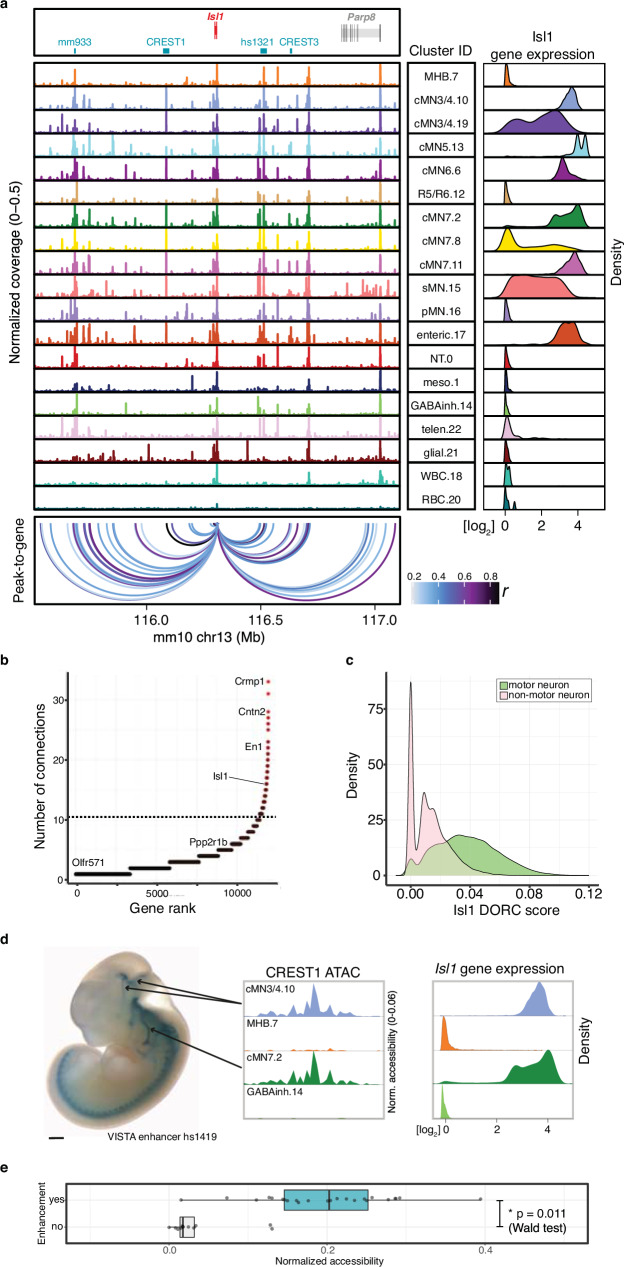
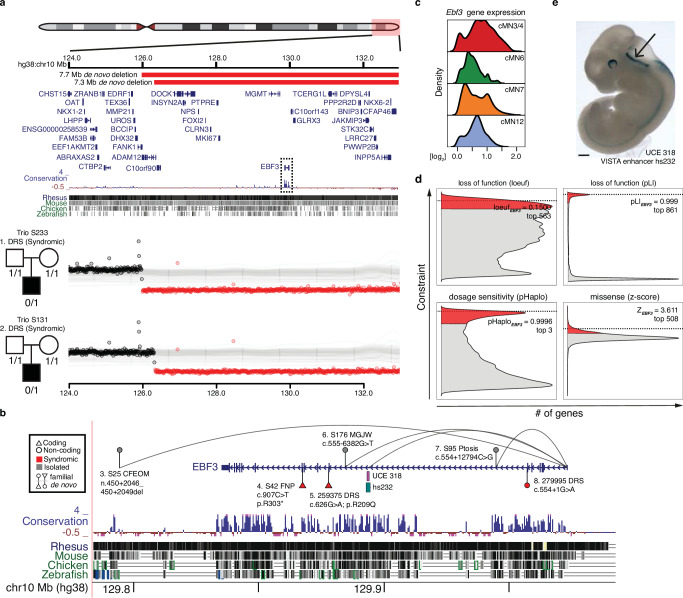
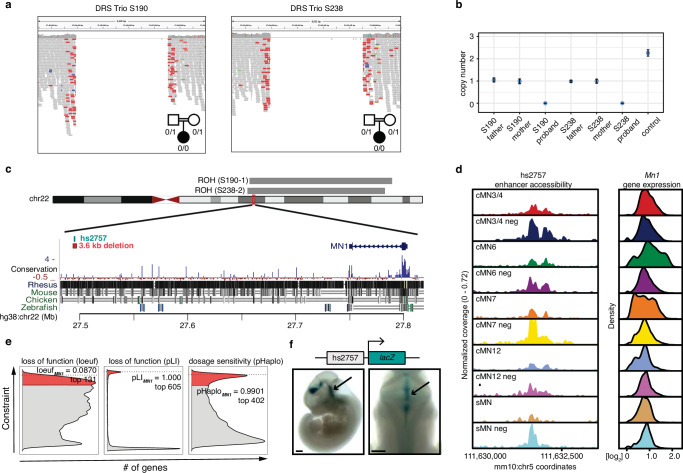
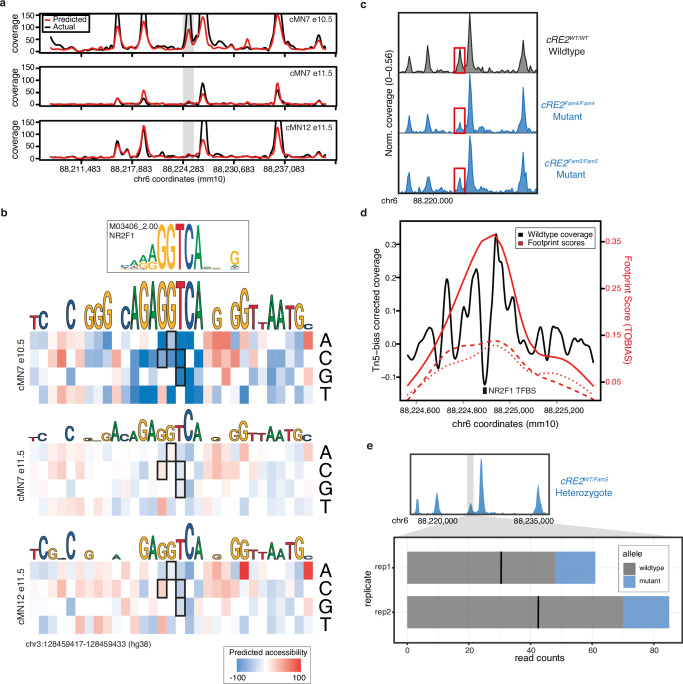
Unsolved Mendelian cases often lack obvious pathogenic coding variants, suggesting potential non-coding etiologies. Here, we present a single cell multi-omic framework integrating embryonic mouse chromatin accessibility, histone modification, and gene expression assays to discover cranial motor neuron (cMN) cis-regulatory elements and subsequently nominate candidate non-coding variants in the congenital cranial dysinnervation disorders (CCDDs), a set of Mendelian disorders altering cMN development. We generate single cell epigenomic profiles for ~86,000 cMNs and related cell types, identifying ~250,000 accessible regulatory elements with cognate gene predictions for ~145,000 putative enhancers. We evaluate enhancer activity for 59 elements using an in vivo transgenic assay and validate 44 (75%), demonstrating that single cell accessibility can be a strong predictor of enhancer activity. Applying our cMN atlas to 899 whole genome sequences from 270 genetically unsolved CCDD pedigrees, we achieve significant reduction in our variant search space and nominate candidate variants predicted to regulate known CCDD disease genes MAFB, PHOX2A, CHN1, and EBF3 - as well as candidates in recurrently mutated enhancers through peak- and gene-centric allelic aggregation. This work delivers non-coding variant discoveries of relevance to CCDDs and a generalizable framework for nominating non-coding variants of potentially high functional impact in other Mendelian disorders.
© 2024. The Author(s).
Conflict of interest statement
D.G.M. is a paid advisor to GlaxoSmithKline, Insitro, and Overtone Therapeutics, and has received research support from AbbVie, Astellas, Biogen, BioMarin, Eisai, Google, Merck, Microsoft, Pfizer, and Sanofi-Genzyme. M.E.T. has received research support and/or reagents from Microsoft, Illumina Inc., Pacific Biosciences, and Ionis Pharmaceuticals. Otherwise, the authors declare that they have no competing interests as defined by Nature Research, or other interests that might be perceived to influence the interpretation of this article.
Figures
Update of
-
A cell type-aware framework for nominating non-coding variants in Mendelian regulatory disorders.medRxiv [Preprint]. 2023 Dec 27:2023.12.22.23300468. doi: 10.1101/2023.12.22.23300468. medRxiv. 2023. Update in: Nat Commun. 2024 Sep 27;15(1):8268. doi: 10.1038/s41467-024-52463-7. PMID: 38234731 Free PMC article. Updated. Preprint.
References
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- figshare/10.25452/figshare.plus.26517577.v1
Grants and funding
- R01 HG003988/HG/NHGRI NIH HHS/United States
- R01 MH115957/MH/NIMH NIH HHS/United States
- DP2 AG072437/AG/NIA NIH HHS/United States
- R01 HD114353/HD/NICHD NIH HHS/United States
- K08 NS099502/NS/NINDS NIH HHS/United States
- R01 EY027421/EY/NEI NIH HHS/United States
- U54 HD090255/HD/NICHD NIH HHS/United States
- T32 NS007473/NS/NINDS NIH HHS/United States
- T32 GM007748/GM/NIGMS NIH HHS/United States
- T32 EY007145/EY/NEI NIH HHS/United States
- P50 HD105351/HD/NICHD NIH HHS/United States
- T32 HL007627/HL/NHLBI NIH HHS/United States
- DE-AC02-05CH11231/Department of Energy
