

ParSE-seq: a calibrated multiplexed assay to facilitate the clinical classification of putative splice-altering variants
- PMID: 39333091
- PMCID: PMC11437130
- DOI: 10.1038/s41467-024-52474-4
ParSE-seq: a calibrated multiplexed assay to facilitate the clinical classification of putative splice-altering variants
Abstract
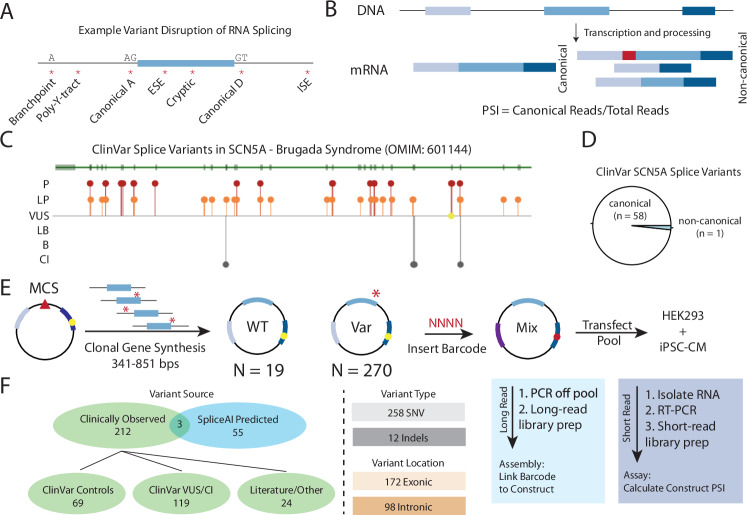
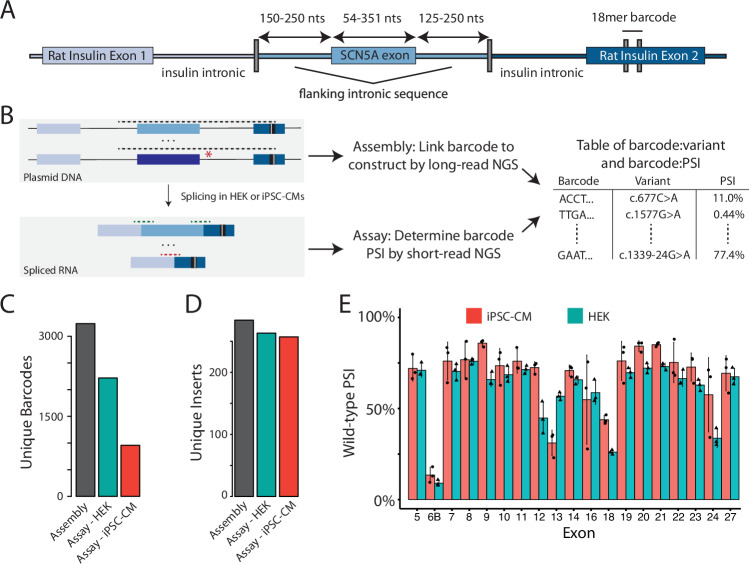
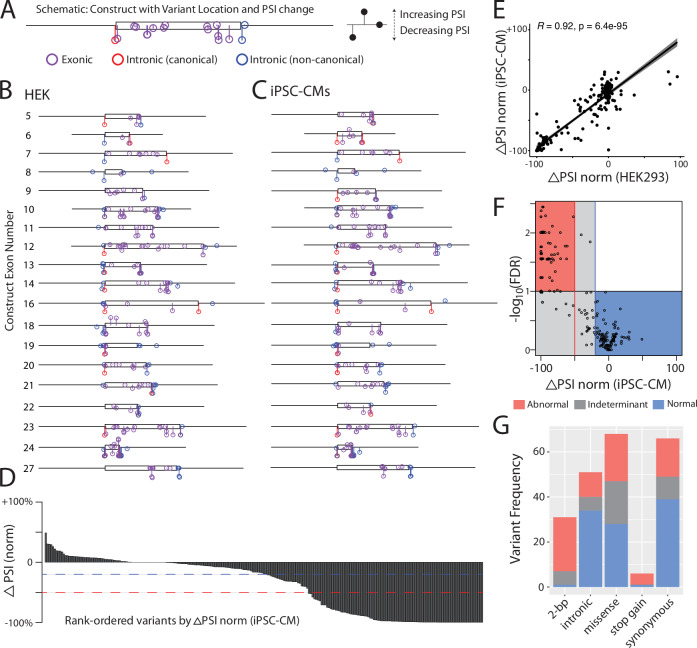
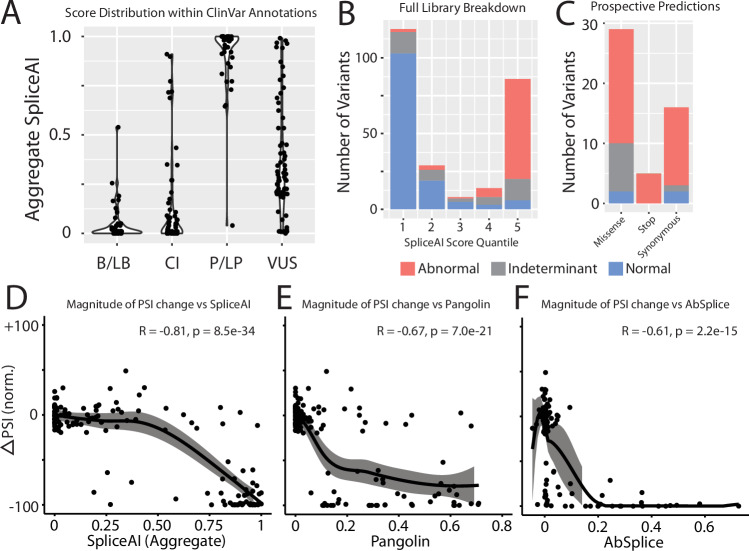
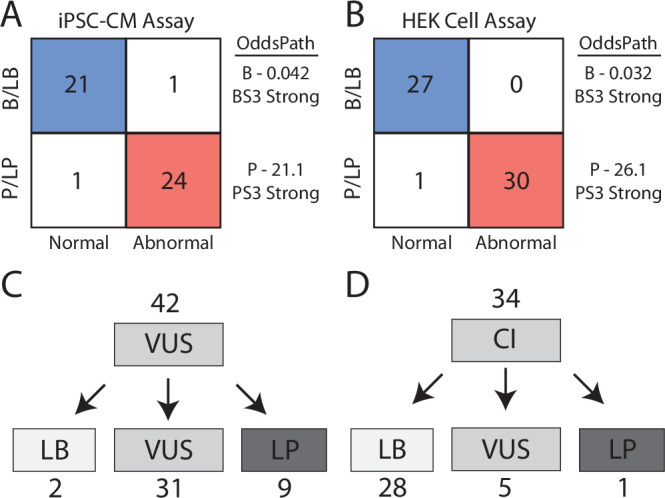
Interpreting the clinical significance of putative splice-altering variants outside canonical splice sites remains difficult without time-intensive experimental studies. To address this, we introduce Parallel Splice Effect Sequencing (ParSE-seq), a multiplexed assay to quantify variant effects on RNA splicing. We first apply this technique to study hundreds of variants in the arrhythmia-associated gene SCN5A. Variants are studied in 'minigene' plasmids with molecular barcodes to allow pooled variant effect quantification. We perform experiments in two cell types, including disease-relevant induced pluripotent stem cell-derived cardiomyocytes (iPSC-CMs). The assay strongly separates known control variants from ClinVar, enabling quantitative calibration of the ParSE-seq assay. Using these evidence strengths and experimental data, we reclassify 29 of 34 variants with conflicting interpretations and 11 of 42 variants of uncertain significance. In addition to intronic variants, we show that many synonymous and missense variants disrupted RNA splicing. Two splice-altering variants in the assay also disrupt splicing and sodium current when introduced into iPSC-CMs by CRISPR-Cas9 editing. ParSE-seq provides high-throughput experimental data for RNA-splicing to support precision medicine efforts and can be readily adopted to study other loss-of-function genotype-phenotype relationships.
© 2024. The Author(s).
Conflict of interest statement
A.M.G. is a consultant for BioMarin, Inc. All remaining authors declare no competing interests.
Figures

Update of
-
ParSE-seq: A Calibrated Multiplexed Assay to Facilitate the Clinical Classification of Putative Splice-altering Variants.medRxiv [Preprint]. 2023 Sep 8:2023.09.04.23295019. doi: 10.1101/2023.09.04.23295019. medRxiv. 2023. Update in: Nat Commun. 2024 Sep 27;15(1):8320. doi: 10.1038/s41467-024-52474-4. PMID: 37732247 Free PMC article. Updated. Preprint.
References
Publication types
MeSH terms
Substances
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
