

Genome-wide impact of codon usage bias on translation optimization in Drosophila melanogaster
- PMID: 39333102
- PMCID: PMC11437122
- DOI: 10.1038/s41467-024-52660-4
Genome-wide impact of codon usage bias on translation optimization in Drosophila melanogaster
Abstract
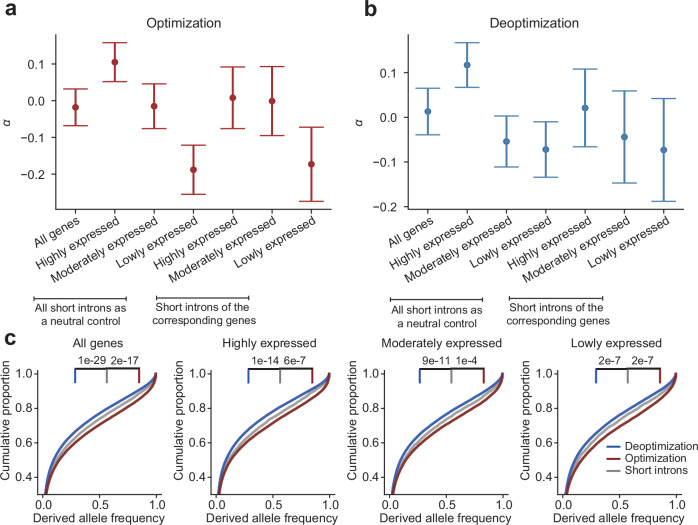
Accuracy and efficiency are fundamental to mRNA translation. Codon usage bias is widespread across species. Despite the long-standing association between optimized codon usage and improved translation, our understanding of its evolutionary basis and functional effects remains limited. Drosophila is widely used to study codon usage bias, but genome-scale experimental data are scarce. Using high-resolution mass spectrometry data from Drosophila melanogaster, we show that optimal codons have lower translation errors than nonoptimal codons after accounting for these biases. Genomic-scale analysis of ribosome profiling data shows that optimal codons are translated more rapidly than nonoptimal codons. Although we find no long-term selection favoring synonymous mutations in D. melanogaster after diverging from D. simulans, we identify signatures of positive selection driving codon optimization in the D. melanogaster population. These findings expand our understanding of the functional consequences of codon optimization and serve as a foundation for future investigations.
© 2024. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
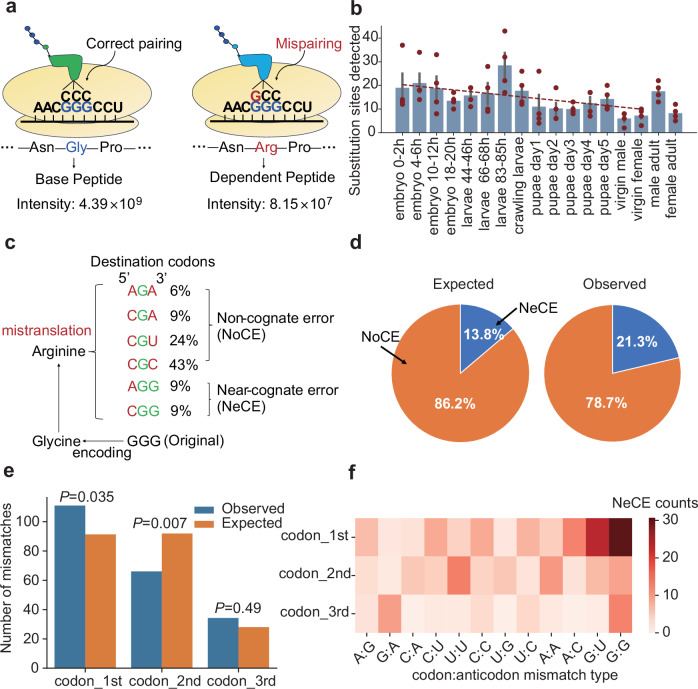
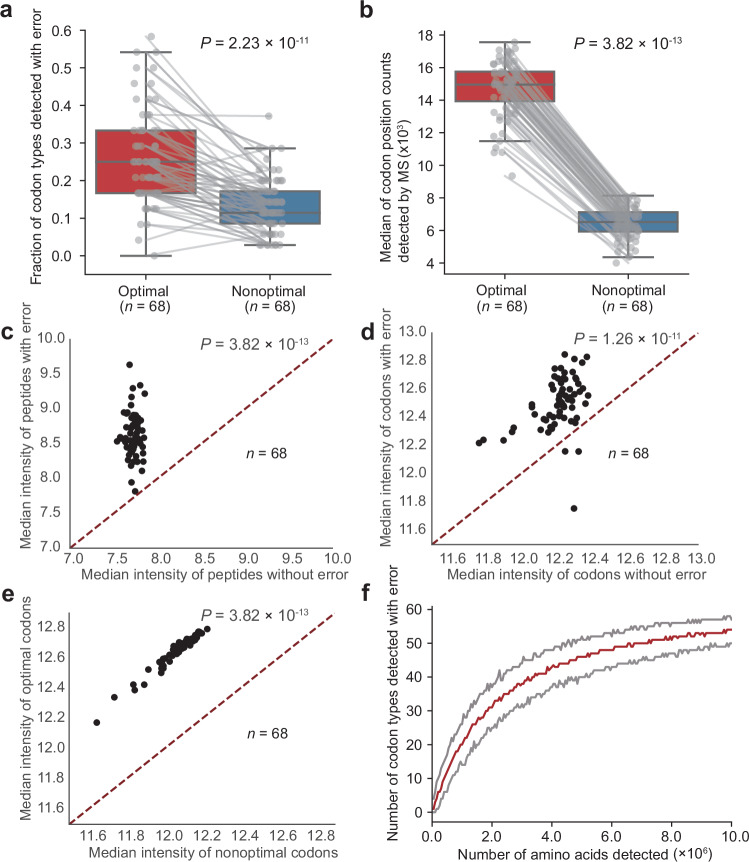
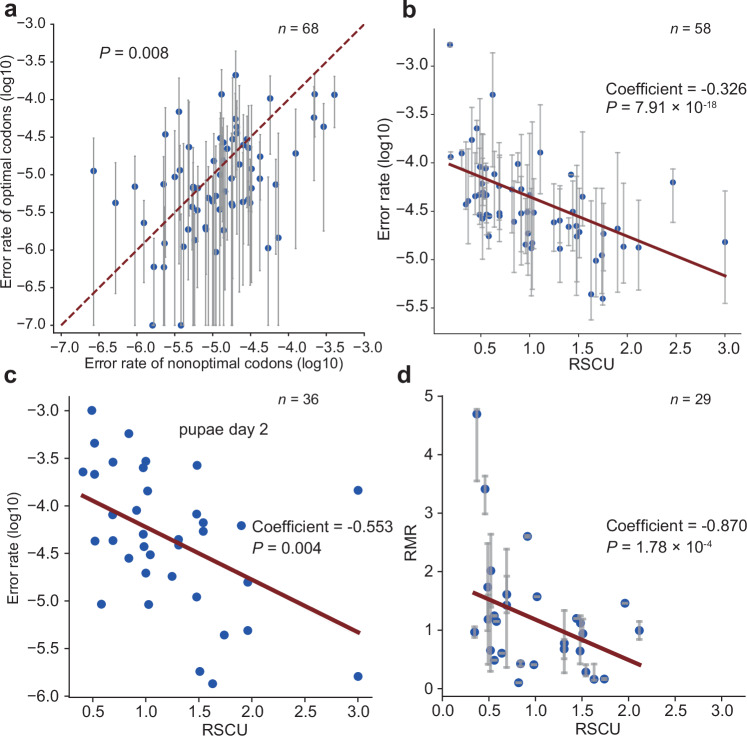
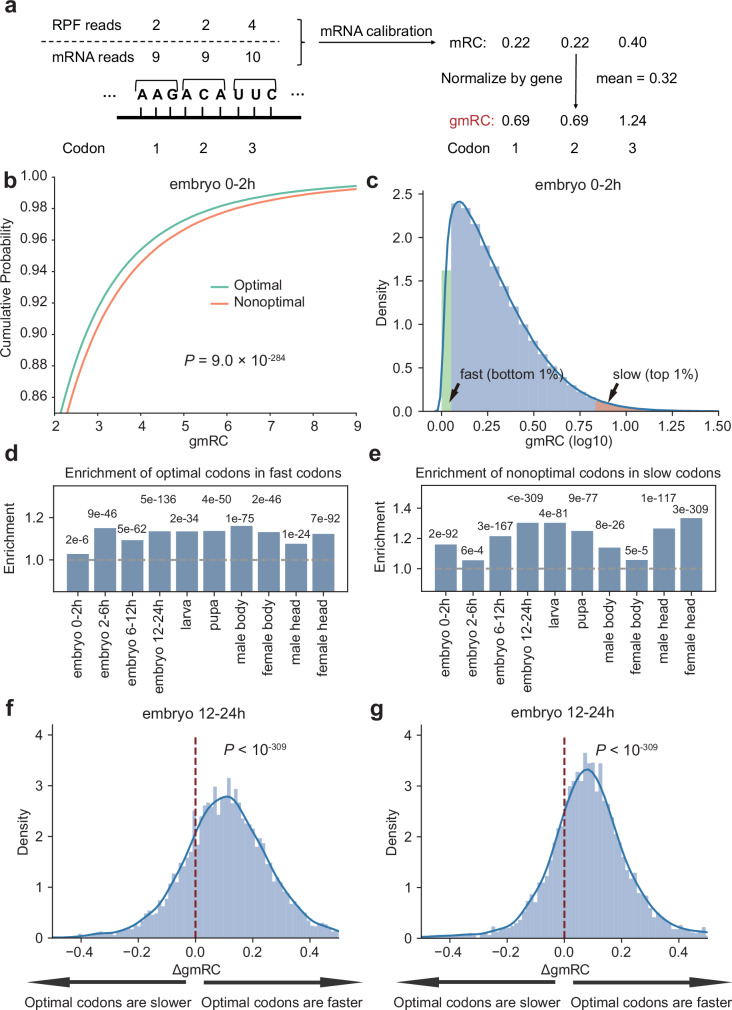
References
Publication types
MeSH terms
Substances
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
